










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.108 n.11-12 Pretoria Jan. 2012
REVIEW ARTICLE
Next generation shotgun sequencing and the challenges of de novo genome assembly
Stephen Schlebusch; Nicola Illing
Department of Molecular and Cell Biology, University of Cape Town, Cape Town, South Africa
ABSTRACT
Sequencing the genomes of the many scientifically fascinating plants and animals found in South Africa is fast becoming a viable option as a result of the rapid and sustained drop in the cost of next generation sequencing over the last five years. However, the processing and assembly of the sequence data produced is not trivial. There are several factors which need to be taken into consideration when planning a strategy to assemble genome sequence data de novo. This paper reviews the advances and the challenges in two of the most rapidly developing areas of the field: the sequencing technology and the software programs used to assemble de novo the sequence data generated by these technologies into a genome.
Introduction
The structure of genomes varies greatly throughout the three domains of life. Eubacterial and archaebacterial genomes are arranged into a single circular chromosome. Eukaryote genomes are arranged into multiple linear chromosomes within the nucleus, and can be made challenging by features such as introns, low complexity repetitive elements, large genome sizes and extensive sequence duplications. These problems are generally more substantial in the plant and animal kingdoms, which tend to have a large number of repetitive regions and are prone to ploidy changes. The sequencing of whole genomes has traditionally been restricted to small genomes or genomes of model organisms which are of particular global interest, because of the high cost of sequencing and the complexity of assembling all the sequence reads. Sequencing of other eukaryote genomes (so called 'non-model organisms'), which may be of biological or evolutionary importance, has to date been considered unfeasible within the resources of the average research laboratory. This scenario is changing.
In the last five years, the development of next generation sequencing technology has seen a major increase in efficiency (the number of base pairs sequenced per day) as well as a vast decrease in cost per base, with prices approximately halving every five months since 2007.1 Unfortunately, the read length (the number of nucleotides sequenced off one fragment) has seen a large decrease compared to that of the original Sanger methods.2 This reduction has posed a major challenge for genome assembly as it makes the definition of areas that have repeated elements for long stretches extremely difficult. The severity of this problem increases with the size of the genome and the number of variable repeat regions. It was for this reason that de novo sequencing of the genomes of complex non-model organisms using solely next generation sequencing technology initially looked dubious.3 Improvements in next generation sequencing technology to increase sequence read lengths, together with the development of new software tools to assemble the sequence reads, has, however, made the sequencing of the genomes of non-model organisms feasible for the first time, as demonstrated recently by the sequencing and assembly of the giant panda4 and strawberry genomes.5
Next generation sequencing technology
The phrase 'next generation sequencing' is a general term applied to sequencing platforms that use post-Sanger technology to sequence large numbers of DNA fragments in parallel.6 Roche, through their '454' sequencing platform, developed the first viable high-throughput sequencing technique.7 The genomic DNA is randomly sheared into fragments of approximately 800 bp to 1000 bp in length, although a shorter fragment works better if maximum read length is not required. Adaptor primer sequences are ligated onto the ends of the genome fragments, which are then hybridised to beads covered in one of the complementary primers. The beads are diluted in an oil emulsion, such that each oil droplet contains only one bead. The DNA fragment attached to each bead is amplified by the polymerase chain reaction (PCR) in a process known as emulsion PCR or emPCR. Each bead is then aliquoted into a welled plate, such that there is only one bead in each well. The actual sequencing commences with the plates undergoing a series of washes. Each wash contains one of the four nucleotides. The incorporation of these nucleotides into a sequence yields a light reaction which can be measured. The number of nucleotides incorporated into a sequence per wash is deduced by the strength of the signal. This method is referred to as pyrosequencing or sequencing by synthesis. The 454 platform, GS FLX+, typically gives a mode read length of 700 bp (Table 1) and is capable of reaching 1 kb. Unfortunately, the 454 platform is not accurate when it comes to sequencing long homogeneous nucleotide runs, resulting in a high chance of a measured insertion or deletion in these regions.
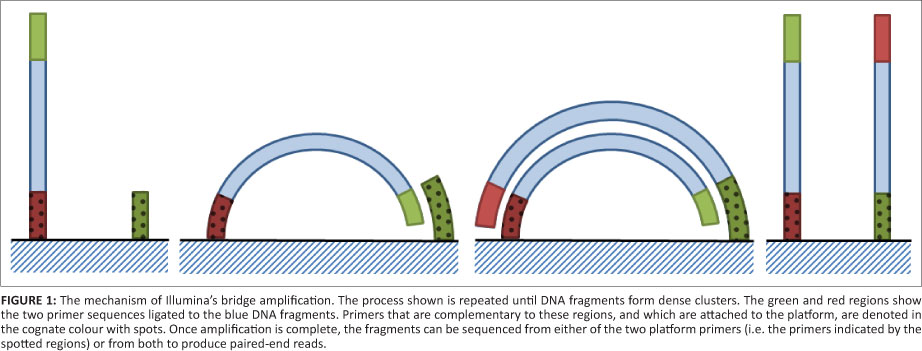
Illumina's platform (originally owned by Solexa) was the next product out on the market. In this method, the genomic DNA is fragmented to a desired size (less than a 1 kb) and adaptors are ligated to either end in a similar manner to Roche's method. These fragments are then chemically attached to a glass slide covered in complementary oligonucleotides of both primers called a flow cell. A method called bridge amplification (Figure 1) is then used to amplify the DNA fragments.8 This amplification results in copies of the original fragment being arranged in clusters around the flow cell. These clusters are then sequenced one nucleotide at a time by washing with a buffer containing fluorescently tagged, reversibly terminating nucleotides.9 The reversibly terminating nucleotides allow all four nucleotides to be washed over at once as only one can be incorporated into the sequence while the fluorescent tag is still attached; each nucleotide has a different colour tag. The colour of each cluster is then recorded, the fluorescent tag removed and the next wash commenced.10 The fragment can be sequenced from both of the primers, resulting in two sequences with a known number of nucleotides between them, depending on the initial fragment size (Table 1). This method is called a paired-end read and can be used to partially compensate for the short read lengths in de novo assembly. Read lengths vary between 100 bp and 150 bp depending on the platform, with a length of 250 bp to be introduced on the MiSeq platform in 2012.
SOLiD sequencing, the third well-established next generation sequencing method, was developed by Applied Biosystems and then later by Life Technologies. The DNA fragments are amplified in a similar manner to that of Roche's system (bead emulsion PCR),11 but the actual sequencing happens in a very different manner. Instead of sequencing by synthesis, SOLiD uses the binding of eight-base oligoprimers to differentiate between nucleotides on the DNA fragment. Although an oligoprimer consists of eight nucleotides, six nucleotides are degenerate, leaving only two that are specific to the query sequence (the two closest to the 3' end; Figure 2a). Thus 16 oligoprimers are supplied in each wash (all possible dinucleotide combinations). The nucleotides closest to the 5' end are marked with one of four fluorescent dyes (with four dinucleotide combinations per colour; Figure 2b).
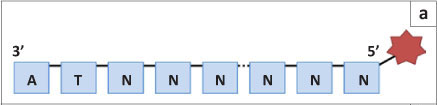
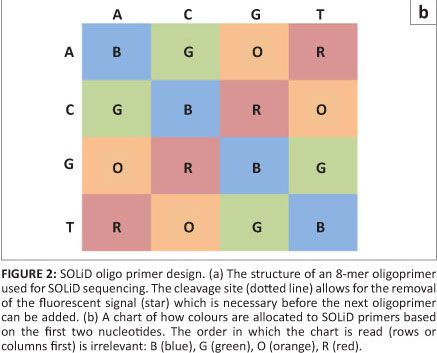
In the first wash of the SOLiD sequencing procedure, the appropriate oligoprimer is ligated to an initial universal primer (n) that is complementary to the primer ligated on the DNA fragment and used for amplification (Figure 3). Once the fluorescent signal is recorded, the last three nucleotides
(which contain the fluorescent dye) are cleaved off and another wash of oligoprimers is applied. This process is repeated until the DNA fragment is completely covered by oligo remnants. The now double-stranded DNA is denatured and the oligoprimer remnants are discarded. The entire process is repeated four more times, but each time with a slightly smaller initial primer (n-1, n-2, etc.). In this way, each nucleotide is analysed twice by dinucleotide probes (Figure 3) and results in a sequence (referred to as colour space) that is directly translatable to genetic code as long as the first nucleotide is known, which it must be because the second sequencing reaction is anchored with an initial universal primer at n-1 (Figure 3). The advantage of this drastic departure from the more conventional sequencing technologies is that it is possible to differentiate between a single nucleotide polymorphism and a sequencing error in the colour space sequence. This ability is because a single nucleotide polymorphism affects two of the colours in the colour space (measured twice), whereas a sequencing error changes only one of the colours. The platform is also capable of paired-end reads, but the second read of each pair is considerably shorter than the primary read (35 bp instead of 75 bp; Table 1). These shorter read lengths make SOLiD's application to the de novo assembly of larger genomes limited.
In addition to SOLiD, Life Technologies have also acquired the rights to a second sequencing technology. Known as Ion Torrent, this platform is very similar to Roche's 454 in many respects. Fragments are amplified by emulsion PCR before being sequenced by applying the nucleotides one at a time and measuring the signal strength emitted.12 What makes Ion Torrent different from the rest is the fact that it measures the change in pH from an incorporated nucleotide rather than a light signal, resulting in very rapid sequencing (Table 1). The measurement of pH change is accomplished using semiconductor chips and an ion-sensitive layer. Read lengths are currently 200 bp, but lengths of 400 bp are expected to be available soon. Unfortunately, because the reaction is not limited to the incorporation of a single nucleotide, it is subject to the same error in regions of homogeneous nucleotide runs that 454 is.
All of the aforementioned technologies amplify the DNA before sequencing. In addition to using time and money, this approach introduces an amplification bias against 'g-c' or 'at' rich areas. However, there are a few sequencing platforms that do not amplify the target DNA. These single-molecule sequencers have been dubbed the 'third generation' (making amplification-dependent sequencers the 'second generation'). Notably among these third-generation sequencers is the PacBio RS from Pacific Bioscience. The PacBio RS rapidly sequences long individual fragments (10 kb) using a system it calls SMRT (single-molecule real-time) technology; (Table 1).12
In this system, DNA polymerases attached to a detection surface are each given a single DNA strand. This strand is then sequenced by measuring the signal given off whenever a phospholinked nucleotide is incorporated into the strand. Each nucleotide is tagged with a different colour which is cleaved when it is added to the existing strand. There is therefore no need for washes or terminating nucleotides. The nucleotide suspension is simply applied to the plate and the sequencing commences. This process therefore results in much longer read lengths than the other currently available platforms, but at a much higher error rate (15%).6 In an effort to try and account for this error rate, in addition to sequencing long single strands, Pacific Bioscience offer a service where they take a shorter piece of double stranded DNA (2 kb for example), and add hairpin adaptors onto the end. This addition has the effect of circularising the DNA, which allows it to be sequenced repeatedly by the DNA polymerase, dramatically reducing the error rate. This is known as circular consensus sequencing. Pacific Bioscience will not be the only sequencer with 10-kb read lengths for long - soon the sequencers GridION and MinION from Oxford Nanopore Technologies will join them.13
For lack of these long read lengths, the original three technologies (Illumina, 454 and SOLiD) are at least able to compensate with the creation of mate-paired reads. Mate-paired reads can be thought of as paired-end reads with a very long gap between them (several kilobases). However, mate-paired reads require a more complicated library construction than do normal genomic reads. With mate-paired reads, the genome is initially fragmented to allow for large gaps of a desired length. These fragments are then circularised with biotinylated primers (Figure 4). The circular DNA is then re-fragmented and the biotinylated regions are purified out, resulting in two sections of DNA that were once separated by kilobases of DNA on the same sequencable fragment. The incorporation of mate-paired reads is required for definition of the longer repetitive elements in a eukaryotic genome and is necessary for analyses into structural variation.

For more information on the chemistry of the different techniques, we recommend the reading of Metzker's12 review and for more information about individual platforms offered by each company and their associated prices, we refer you to Glenn6.
De novo genome assembly
Once the genome has been sequenced, the millions of short sequence reads need to be assembled into an informative model of the genome. However, the extremely large number of reads can make the computation very taxing. A good assembly algorithm therefore needs to be as efficient as possible within a computer's processing power, without sacrificing too much accuracy.
The compromise between speed and accuracy is commonly helped through the creation of k-mers.14 K-mers are created by breaking down the existing sequences into shorter sequences of length 'k'. These allow the data to be summarised and compacted in such a way that commonalities can be identified and links drawn between sequences. K-mer information is used to varying degrees in different algorithm strategies and programs. Overlap assembly strategies such as Greedy and Overlap/Layout/Consensus use k-mer data to efficiently find the best overlap amongst the sequences, whereas Eulerian strategies use the k-mers to create a de Bruijn graph.15
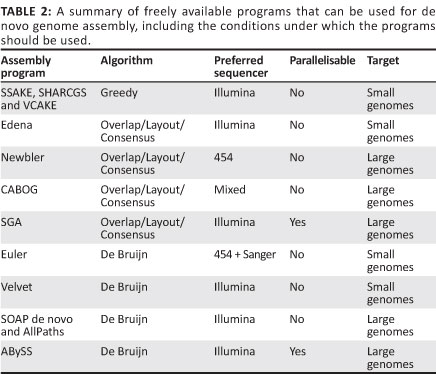
Greedy algorithm
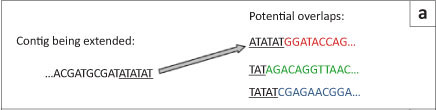
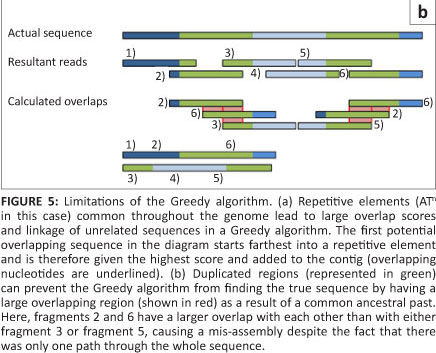
The first short-read genome assembly programs (for example SSAKE,16 SHARCGS17 and VCAKE18) were based on the Greedy algorithm (Table 2).14 This algorithm works in an intuitive manner by finding high scoring overlap sequences and joining them together. In this way, contigs are produced and extended. However, the localised view that this procedure maximises, opens it up to several pitfalls. For example, repetitive elements within the genome, if not treated with care, can cause hybrid contigs to be formed (Figure 5a).3 To address this issue, most Greedy algorithms either stop extension if several good hits are found that do not agree with each other (i.e. they do not share sufficient commonalities) or do not take into account elements in the overlap that are overly represented within the data set (suggesting it is found multiple times in the genome).15 The local maximisation also tends to lead to solutions that are not globally optimal (Figure 5b). This tendency to get caught in local maxima limits the use of this algorithm to small simple genomes.19
Overlap/Layout/Consensus algorithm
The Overlap/Layout/Consensus method is a three-step process that effectively combines the overlap idea of the Greedy algorithms with a global view point to prevent local maxima from being pursued (an Overlap/Layout/ Consensus algorithm would have successfully put together the correct sequence in Figure 5b for example).15 The first step of the method identifies overlaps between all the sequences. This pairwise alignment, while streamlined with the use of indexing and k-mers, can still be computationally expensive in larger data sets. The sequences are then mapped out on a graph as nodes and connected according to the overlap information (layout step). This step is kept computationally efficient as actual sequence composition is not considered.14 Ideally a single path, called the Hamiltonian path, is identified that goes through all the sequences exactly once (or rather one path per chromosome). In reality, however, this single path is prevented by insufficient coverage, sequencing errors, genetic polymorphism and unresolvable elements caused by repeated elements. Once the optimum path has been derived, sequence identity is decided by compiling evidence for each nucleotide in the consensus step. Examples of Overlap/Layout/Consensus programs are Newbler,7 Edena20 and CABOG21 (Table 2).
The computational requirements of this strategy are proportional to the number of sequence reads in the data set. These programs have therefore not done well with the exponential increase in next generation sequencing data. While programs like Newbler and CABOG have been used to assemble large genomes, these assemblies were supplemented by long Sanger reads. Such programs would struggle to scale up to the hundreds of millions of reads that would be necessary for a large de novo assembly consisting purely of next generation sequencing data.19 The strategy should not, however, be dismissed yet. A new take on the method, called SGA (String Graph Assembly), was recently developed by Simpson and Durbin22; SGA uses a highly effective compression technique that allows for the assembly of genomes with minimal RAM usage.
De Bruijn algorithm
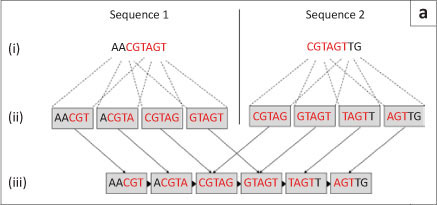
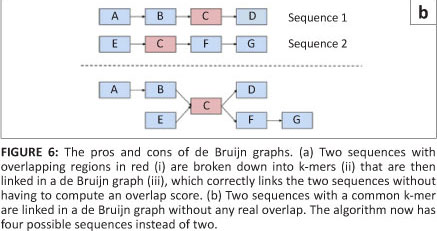
The second type of graph assembly technique is based on the formation of de Bruijn graphs (also known as an Eulerian algorithm).23 These graphs are formed from the creation of k-mers of the original sequences. These k-mers are then connected to k-mers that they overlap with at k-1 sites. In this way, the algorithm avoids the computationally difficult global pairwise overlap step used in the Greedy and Overlap/Layout/Consensus strategies as the overlap is implicit (Figure 6a). However, the conversion of sequences into k-mers does result in the loss of some information. This loss can cause the algorithm to create links between two unrelated sequences that it then cannot dismantle again without consulting the original reads (Figure 6b).14 The larger the value of 'k' and the smaller the genome, the fewer false associations will be made in the de Bruijn graph. The value of 'k' cannot be made too large, however, as sequences still need to share 'k-1' nucleotides in the real overlap in order to be associated.
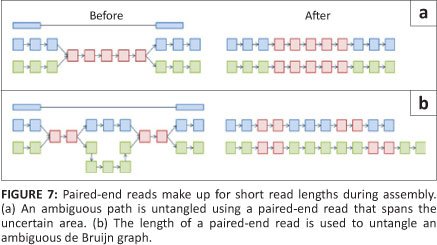
Once the complete de Bruijn graph has been created from the k-mers, a path that uses every node (called the Eulerian path) must be drawn through the system. As a consequence of collapsed nodes (as seen in Figure 6b), there are normally several possible paths through the graph. In order to separate the true path from the false ones, sequence information needs to be added back into the network (Figure 7).14 Once the best path has been identified, the contig sequence can be read from the k-mer identities. Examples of de Bruijn graph programs are Euler,23 Velvet,24 ABySS,25 AllPaths26 and SOAP de novo27 (Table 2). These programs are appealing because, theoretically, the computational requirements increase with genome size and not with the number of reads. The fact that computational requirements are independent of the number of reads is important because (without sequencing error) a genome will be broken down into the same de Bruijn graph, regardless of the coverage. Of these programs, Allpaths LG and SOAP de novo have been designed specifically to deal with large genomes and were ranked first and second, respectively, in an open assembly competition hosted by an independent third party, with SGA being placed third.28 SOAP de novo was also the program used to assemble the giant panda genome.
Dealing with sequence errors
Many of the problems faced by genome assembly algorithms, particularly those using a de Bruijn graph, are convoluted by the existence of sequencing errors.14 Although low, different technologies have different error rates (reported on their respective websites). For example, SOLiD sequencing has an initial accuracy of 99.94% for each nucleotide. This accuracy is reduced to 99% by the 35th base pair. In comparison, the 454 sequencer's accuracy decreases to 99% only at the 400th base pair. Reports on the Illumina platform indicate that 80% of nucleotides are 99.9% accurate. Even with these levels of accuracy, sequencing a genome will result in anywhere between hundreds of thousands to tens of millions of incorrect bases. The probability of any one nucleotide being incorrect is reported with each call by the sequencer. Although this information is useful for determining the overall quality of a sequencing run, the inclusion of it in the actual genome assembly is a computationally costly endeavour.19 Because of the cost, most assembly programs choose to ignore data that include the probability of the error (e.g. Edena, Velvet, SOAP de novo and SSAKE).
Theoretically, Greedy algorithms are well protected from errors in a high-coverage situation as sequences without error should be incorporated preferentially to those with an error (because of a higher overlap score). Overlap algorithms in general also avoid using error probabilities by not requiring a 100% sequence similarity, allowing the error to be passed over and resolved later in the consensus step. The de Bruijn graph method on the other hand, does not have one of these inherent mechanisms to deal with sequence errors.14 If unaddressed, sequence errors can cause excessive complexity in the de Bruijn graph by adding so-called 'bubbles' (which occur when an otherwise linear path has two possible central sequences) and 'spurs' (which occur when a sequence has two possible starts or ends) to an otherwise unambiguous path (Figure 8) or by causing collapsed nodes (as in Figure 6b). The creation of bubbles and spurs is the most common outcome of an error as 4k should be much larger than the genome, meaning novel k-mer creation is the most likely outcome of an incorrect base. The creation of novel k-mers makes sequence errors especially troublesome in de Bruijn graphs, as one error can potentially result in 'k' new k-mers in a graph. However, not all bubbles and spurs are the result of sequencing errors; bubbles and spurs can also be caused by single nucleotide polymorphisms, microsatellites and tandemly duplicated areas.
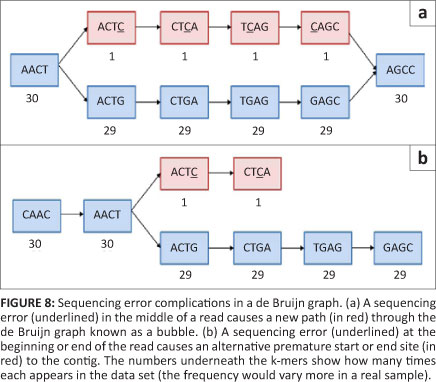
Methods for dealing with errors normally revolve around this concept of original k-mer creation. The Euler method, for example, plots the k-mers versus their frequency in the data,23 creating a bimodal graph, with one peak representing the real k-mers observed and a second peak (lower on the graph) representing the new k-mers created by sequencing errors. A point between these modes is chosen and everything above this point is trusted and everything below the point is distrusted. A similar mechanism is employed in many programs. Suspicious k-mers can either be altered to a more likely k-mer (Euler) or discarded (Velvet). This approach does, however, mean that true k-mers with a very low coverage will be adjusted or disregarded and certain errors that happened to result in a common k-mer will be accepted. Once a graph has been assembled, there are several simplification algorithms that trim spurs and collapse bubbles to make informative contigs.14 This necessary step does unfortunately result in a loss of real information like single nucleotide polymorphisms and occasionally even entire exons.29
Conclusion
Given their differences, the question arises as to which assembler and sequencing platform should be used under which conditions. This question is not an easy one to answer. The main variables to consider are the budget for sequencing, the computer processing power and the genome size. The decision is made more difficult by the fact that sequencer technology does not stand still - new assembly programs are constantly being released and old programs are being updated. Generally though:
- SOLiD's advantage of being able to identify single nucleotide polymorphisms does not make up for its shorter read length which makes the de novo assembly of complex genomes difficult.
- Sequencing on Roche's 454 is expensive, but it has a good read length, which is especially important for the de novo assembly of complex genomes.
- Pacific Bioscience has surpassed 454 in read length for a similar cost.
- Illumina's platform provides a good balance between read length and cost; and is the most widely used for such projects.
- Paired-end reads on the Illumina or SOLiD platforms are worth the extra cost.
- Ion Torrent offers fast sequencing turnover combined with a reasonable read length; however, the lack of paired-end reads limits it to smaller genomes.
- Greedy algorithms should not be used unless computer processing power is limited and the genome is simple.
- With the exception of SGA, the de Bruijn graph programs cope better with the large numbers of short reads that Illumina or SOLiD will produce.
- Having a portion of your coverage as either mate-paired reads or long third-generation reads will significantly decrease the fragmentation of the final assembly.
- Extreme guanidine-cytosine contents can affect the amplification efficiency of DNA and thus negatively impact second-generation sequencing.
- Check to see whether a potential reference genome exists before attempting de novo assembly (http://www.ncbi.nlm.nih.gov/genome/browse/).
If you can afford it, a good strategy is to combine data from two different platforms (for example Illumina and Pacific Bioscience), which would compensate for each method's deficiencies. This strategy gives the best chance of a complete and accurate genome description. However, even under the best conditions, the de novo assembly of large genomes will be fractured and incomplete to some degree. Nevertheless, this deficiency should not deter researchers from taking advantage of these new sequencing technologies to assemble the genomes of diverse plants and animals, even if they are incomplete, to answer important biological and evolutionary questions. Genome sequences, even fractured ones, are a wealth of potential information.
Acknowledgements
This work was funded by the National Research Foundation (South Africa) and the University of Cape Town.
Competing interests
We declare that we have no financial or personal relationships which may have inappropriately influenced us in writing this paper.
Authors' contributions
N.I. conceived the scope of the review. S.S. wrote the first draft of the manuscript. N.I. and S.S. worked together on subsequent versions of the manuscript.
References
1. Stein LD. The case for cloud computing in genome informatics. Genome Biol. 2010;11:207. http://dx.doi.org/10.1186/gb-2010-11-5-207 [ Links ]
2. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;74(12):5463-5467. http://dx.doi.org/10.1073/pnas.74.12.5463 [ Links ]
3. Pop M, Salzberg SL. Bioinformatics challenges of new sequencing technology. Trends Genet. 2008;24(3):142-149. http://dx.doi.org/10.1016/j.tig.2007.12.006 [ Links ]
4. Li R, Fan W, Tian G, et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463:311-317. http://dx.doi.org/10.1038/nature08696 [ Links ]
5. Shulaev V, Sargent DJ, Crowhurst RN, et al. The genome of woodland strawberry (Fragaria vesca). Nat Genet. 2011;43:109-116. http://dx.doi.org/10.1038/ng.740 [ Links ]
6. Glenn TC. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 2011;11:759-769. http://dx.doi.org/10.1111/j.1755-0998.2011.03024.x [ Links ]
7. Margulies M, Egholm M, Altman WE, et al. Genome sequencing in micro fabricated high-density picolitre reactors. Nature. 2005;437:376-380. [ Links ]
8. Fedurco M, Romieu A, Williams S, Lawrence I, Turcatti G. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucleic Acids Res. 2006;34(3):e22. http://dx.doi.org/10.1093/nar/gnj023 [ Links ]
9. Turcatti G, Romieu A, Fedurco M, Tairi A-P. A new class of cleavable fluorescent nucleotides: Synthesis and optimization as reversible terminators for DNA sequencing by synthesis. Nucleic Acids Res. 2008;36(4):e25. http://dx.doi.org/10.1093/nar/gkn021 [ Links ]
10. Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53-59. http://dx.doi.org/10.1038/nature07517 [ Links ]
11. Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135-1145. http://dx.doi.org/10.1038/nbt1486 [ Links ]
12. Metzker ML. Sequencing technologies - The next generation. Nat Rev Genet. 2010;11:31-46. http://dx.doi.org/10.1038/nrg2626 [ Links ]
13. Lieberman KR, Cherf GM, Doody MJ, Olasagasti F, Kolodji Y, Akeson M. Processive replication of single DNA molecules in a nanopore catalyzed by phi29 DNA polymerase. J Am Chem Soc. 2010;132(50):17961-17972. http://dx.doi.org/10.1021/ja1087612 [ Links ]
14. Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95:315-327. http://dx.doi.org/10.1016/j.ygeno.2010.03.001 [ Links ]
15. Pop M. Genome assembly reborn: Recent computational challenges. Brief Bioinform. 2009;10(4):354-366. http://dx.doi.org/10.1093/bib/bbp026 [ Links ]
16. Warren RL, Sutton GG, Jones SJM, Holt RA. Assembling millions of short DNA sequences using SSAKE. Bioinformatics. 2007;23(4):500-501. http://dx.doi.org/10.1093/bioinformatics/btl629 [ Links ]
17. Dohm JC, Lottaz C, Borodina T, Himmelbauer H. SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing. Genome Res. 2007;17:1697-1706. http://dx.doi.org/10.1101/gr.6435207 [ Links ]
18. Jeck WR, Reinhardt JA, Baltrus DA, et al. Extending assembly of short DNA sequences to handle error. Bioinformatics. 2007;23(21):2942-2944. http://dx.doi.org/10.1093/bioinformatics/btm451 [ Links ]
19. Zhang W, Chen J, Yang Y, Tang Y, Shang J, Shen B. A practical comparison of de novo genome assembly software tools for next-generation sequencing technologies. PLoS One. 2011;6(3):e17915. http://dx.doi.org/10.1371/journal.pone.0017915 [ Links ]
20. Hernandez D, François P, Farinelli L, 0steras M, Schrenzel J. De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802-809. http://dx.doi.org/10.1101/gr.072033.107 [ Links ]
21. Miller JR, Delcher AL, Koren S, et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24(24):2818-2824. http://dx.doi.org/10.1093/bioinformatics/btn548 [ Links ]
22. Simpson JT, Durbin R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 2012;22:549-556. http://dx.doi.org/10.1101/gr.126953.111 [ Links ]
23. Pevzner PA, Tang H, Waterman MS. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci. 2001;98(17):9748-9753. http://dx.doi.org/10.1073/pnas.171285098 [ Links ]
24. Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821-829. http://dx.doi.org/10.1101/gr.074492.107 [ Links ]
25. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol i. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117-1123. http://dx.doi.org/10.1101/gr.089532.108 [ Links ]
26. Butler J, MacCallum I, Kleber M, et al. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810-820. http://dx.doi.org/10.1101/gr.7337908 [ Links ]
27. Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265-272. http://dx.doi.org/10.1101/gr.097261.109 [ Links ]
28. Earl D, Bradnam K, John JS, et al. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011;21:2224-2241. http://dx.doi.org/10.1101/gr.126599.111 [ Links ]
29. Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8:61-65. http://dx.doi.org/10.1038/nmeth.1527 [ Links ]
![]() Correspondence to:
Correspondence to:
Nicola Illing
Department of Molecular and Cell Biology, University of Cape Town, Rondebosch 7700, South Africa
Email: nicola.illing@uct.ac.za
Received: 03 May 2012
Accepted: 21 June 2012
Published: 31 Oct. 2012
© 2012. The Authors. Licensee: AOSIS OpenJournals. This work is licensed under the Creative Commons Attribution License.


{kind=link}
{kind=link}
{kind=link}