










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.108 n.9-10 Pretoria Jan. 2012
REVIEW ARTICLE
Cooperation and conflict in cancer: an evolutionary perspective
Jonathan FeatherstonI, II; Pierre M. DurandI, II
IDepartment of Molecular Medicine and Haematology, University of the Witwatersrand, Johannesburg, South Africa
IINational Health Laboratory Services, Johannesburg, South Africa
ABSTRACT
Evolutionary approaches to carcinogenesis have gained prominence in the literature and enhanced our understanding of cancer. However, an appreciation of neoplasia in the context of evolutionary transitions, particularly the transition from independent genes to a fully integrated genome, is largely absent. In the gene-genome evolutionary transition, mobile genetic elements (MGEs) can be studied as the extant exemplars of selfish autonomous lower-level units that cooperated to form a higher-level, functionally integrated genome. Here, we discuss levels of selection in cancer cells. In particular, we examine the tension between gene and genome units of selection by examining the expression profiles of MGE domains in an array of human cancers. Overall, across diverse cancers, there is an aberrant expression of several families of mobile elements, including the most common MGE in the human genome, retrotransposon LINE 1. These results indicate an alternative life-history strategy for MGEs in the cancers studied. Whether the aberrant expression is the cause or effect of tumourigenesis is unknown, although some evidence suggests that dysregulation of MGEs can play a role in cancer origin and progression. These data are interpreted in combination with phylostratigraphic reports correlating the origin of cancer genes with multicellularity and other potential increases in complexity in cancer cell populations. Cooperation and conflict between individuals at the gene, genome and cell level provide an evolutionary medicine perspective of cancer that enhances our understanding of disease pathogenesis and treatment.
Introduction
Over the last few decades, evolutionary and ecological perspectives of carcinogenesis have become more prominent in the literature. Cancer cells fulfil the criteria for Darwinian evolution by natural selection,1 that is heritable variation in fitness, and investigating neoplasia in this context has generated new insights into disease aetiology, pathogenesis and treatment.2,3,4 Such an application is exemplified by the analysis of Nagy5 who deduced that if heterogeneity is present between cancerous cells then competition for resources (such as vascular nutrients) may occur between tumour cells, leading to 'hypertumours' with a more aggressive phenotype. By this reasoning, cancerous cells may compete with host tissue as well as other cancer cells or groups of cancer cells. Of course, as a result of phenotypic variation, slower-growing, less virulent tumours still exist.
While a number of evolutionary biology and genome-centric concepts have been applied in the study of cancer,6,7,8 an understanding of cancer in terms of evolutionary transitions, particularly the gene-genome transition, is largely absent. Here we discuss cancer from the perspective of evolutionary transitions, highlighting the dynamics of cooperation and conflict that exist between different levels of organisation, with particular interest in the gene-genome transition. Understanding these dynamics provides an enhanced theoretical framework for cancer biology. For cancer research, evolutionary transitions and the closely allied subject of multilevel selection theory have particular relevance and potential applications, including modifying treatment regimens4,9 to account for evolutionary processes and modelling the formation of malignancy in benign tumours.10 Conflict between higher and lower levels of selection, for example the unicellular-multicellular levels of selection, is central to understanding the pathogenesis and evolution of cancer. This importance is highlighted by the finding that the origin of most 'gatekeeper' genes can be traced to the emergence of multicellularity.11 Gatekeeper genes mitigate cooperation and conflict between cells and may include both tumour suppressors and oncogenes. Such conflict can also exist at the gene-genome juncture where selection for mobile genetic elements at the gene level has been hypothesised to play a role in the aetiology and progression of certain cancers.12
Multilevel selection theory, evolutionary transitions and cancer
Multilevel selection theory (MLST), which includes group selection and for which there is now a significant body of evidence, describes the living world in terms of hierarchically structured levels where the tenets of selection are applicable to evolutionary units at varying levels of complexity.13,14,15 Groups of genes form genomes, which form cells, which form multicellular organisms, which may form social groups and so on. Natural selection may act at any of these levels and the fitness conferred by a particular trait can be influenced by selection operating at any level. For example, an expressed trait may be beneficial at one level but potentially harmful at another. A key feature of MLST is that individual units at one level can interact to generate properties at a higher level, which may simply be aggregates of the properties at the lower level or may represent new, emergent properties. Emergent properties are not simply aggregates of the property at the lower level and are not necessarily reducible to them.16 They emerge by interactions within the group and have the ability to create new levels of biological organisation and information leading to a new type of individual at the higher level. In this way, a hierarchical system is generated with higher levels of complexity being derived from lower ones. Evolutionary transition theory refers to these jumps in complexity and attempts to explain why and how these different levels emerge.15,17,18 A common thread in the emergence of the higher level as a discrete evolutionary unit or individual, is that units capable of independent reproduction before the transition are only capable of replicating themselves as part of the higher-level individual after the transition,17 the premier example of which is the transition to multicellularity.19
For the evolutionary biology of cancer, MLST and the transition to multicellularity hold particular relevance. Multicellular organisms comprise populations of cells, which cooperate to enhance the fitness of the whole. However, as with all evolutionary transitions, there is the ever-present temptation to cheat so that fitness at the lower level is enhanced at the expense of the higher level.20 'Cheaters' abort their altruistic behaviour at the higher level in favour of selfish behaviour and a fitness benefit at the lower level. This process is the basic pathology of cancer, albeit that the benefit to cheaters may be transient given that the lower-level unit's survival is usually dependent upon the survival of the higher-level unit (although there are at least two exceptions to this norm, see Murgia et al.21 and Siddle et al.22). Increased fitness at the level of individual cancer cells facilitates clonal evolution and disease progression and decreases the fitness of the organism4 leading to a transfer of fitness to a lower, unicellular level of selection.23,24 In effect, cancer is a failure of cooperation at the cellular level. Treatment strategies for cancer need to be informed by evolutionary theory in order to avoid drug resistance, which forms readily in cancers as a consequence of chemotherapeutic agents essentially selecting for the fitter, drug-resistant cells. Targeting higher-level units (e.g. populations of cells or genomes) rather than lower-level units can be very effective at reducing the rate of drug-resistant mutations.3,4 For example, the use of anti-angiogenic compounds targets the 'group' as opposed to individual cells and results in a major therapeutic benefit. Anti-angiogenic compounds deprive a growing tumour of the rich blood supply needed for rapid growth, which leads to death at a population level where resistance is far less likely to emerge (see Pepper et al.3, Crespi and Summers4 and Durand and Coetzer25).
It has long been argued that cancer and multicellularity are evolutionarily connected.26 Additional evidence for this connection came to light using a method known as phylostratigraphy, which correlates the origin of genes with evolutionary transitions.27 A phylostratigraphic analysis of the conserved domains in cancer genes traced their origin to two major evolutionary events: the emergence of a stable genome and multicellularity.11 The so-called 'gatekeeper' cancer genes are directly connected to the origin of multicellularity in metazoa and the correlation between the emergence of these genes and metazoan multicellularity suggests they played a key role in this transition. Mutations in the same genes disrupt cell signalling and growth functions during carcinogenesis.
The phylostratigraphic analyses also found that genes implicated in genome stability, referred to as 'caretaker' genes, emerged during the origin of the first cell.11 In terms of levels of information organisation, structures such as the cell are not necessarily levels of complexity.14 Such is the case if one subscribes to the view that levels of complexity are about levels of information organisation,17 although this is not universally shared (for example see Griesemer14 for a discussion on replicators versus reproducers). So in this case the level of complexity in question is the genome. In keeping with the genome-centric view of evolution,7 this level is the functional hereditary level at which biological information and complexity reside. Therefore what is significant here is that the emergence of caretaker genes involved in genome maintenance and stability coincide with the origin of the most ancient, functionally integrated genomes.
In addition to the unicellular-multicellular evolutionary transition we consider that other evolutionary transitions, such as the gene-genome evolutionary transition, may be associated with cancer. For example, the caretaker genes, which originate from the early origin of the genome, are less effective in a cancer cell, which results in conflict, or at least increased tension, between gene and genome levels. The genome is the playground for selfish, cooperative and altruistic genes, where tensions between two levels of selection (gene and genome) can arise.8,28 These observations are relevant for our understanding of the gene-genome evolutionary transition.
Mobile genetic elements and cancer
Mobile genetic elements (MGEs) have played a major role in genome evolution by generating new regulatory sites and providing coding material for novel functions. MGEs usually become 'domesticated' by ceasing to exist as individuals with a unique life-history strategy and are incorporated into the higher-level unit of selection - the genome.29 Intragenomic conflict can arise if MGEs are not domesticated.30 MGEs are functionally analogous to the presumed ancient replicators that cooperated to form primitive protein coding genomes and are helpful for understanding the gene-genome evolutionary transition.28 A causal link between MGEs and carcinogenesis has also been identified as MGE activity may cause genome damage,12,31,32,33 dysregulation of genome replication or cell cycling, disruption of cooperative cellular behaviour and eventually neoplasia.34,35,36,37,38 In a recent whole-genome analysis of RNA transcripts using digital gene expression analysis, an increase in satellite repeat marker expression was identified in pancreatic (human and mouse), prostate, lung, kidney and ovarian tumours.39,40 Of the satellite regions expressed in tumours, MGEs, including LINE-1 (the most abundant MGE in the human genome) were highly enriched. The authors of these findings suggest that the aberrant expression of selfish genes was because of epigenetic modification, but they do not state whether the change in expression is causal in the development of cancers or is simply a by-product of other disruptive processes. This phenomenon was found in at least five tumours; however, whether this can be generalised to all cancers is unknown.39 These empirical data of altered MGE activity in various cancers provide an opportunity to analyse the life-history strategy of individual genes in the context of the gene and genome levels of selection in cancer cells. The current review does not attempt to demonstrate that MGEs initiate cancers, although there are some instances where this may be the case, for example, Lamprecht et al.35 found that aberrant activation of the endogenous long terminal repeat (LTR) promoter of retrotransposon THE1B causes overexpression of cell cycling genes and malignant transformation. Here we merely ask whether the life-history strategy of MGEs is altered in cancer and if this change in life-history strategy represents a shift in emphasis from the genome to the gene. Interpreting Lamprecht et al.'s35 findings from the perspective of the gene-genome evolutionary dynamic, one can conclude that unregulated activity of a selfish lower-level unit (THE1B) disrupts homeostasis at the higher level (genome) and induces progression to carcinogenesis. In the ensuing cancer cell population there was further disruption of genegenome homeostasis and, just as in the aberrant unicellular-multicellular dynamic, there was a relative transfer of fitness to the level of the gene.
As described above, the essence of cancer is that there is a disconnection between levels of complexity, the unicellular and multicellular levels, and/or the gene-genome levels. In malignancy, the increasing conflict and decreasing cooperation revolve around these two major evolutionary transitions. The re-organisation of fitness to the individual cell reflects conflict at the unicellular-multicellular bridge as reflected by the discovery that gatekeeper cancer genes arose in tandem with multicellularity.11 As in all biological systems where there is phenotypic variability, such conflict may not be a total breakdown of the unicellular-multicellular transition but rather a heightening of tension between the two levels. Selection operating at the level of the cell in cancer represents a shift from a higher-level unit to a lower-level unit; however, this shift is not a complete disconnect given that the fitness of the higher level is still affected by the fitness of the cancerous cell and increased cellular fitness may lead to the demise of the higher-level unit. Therefore, in cancer, even though conflict between the levels of selection is increased, viability at one level of selection can influence viability at another level.
Is mobile genetic element behaviour disrupted in all cancers?
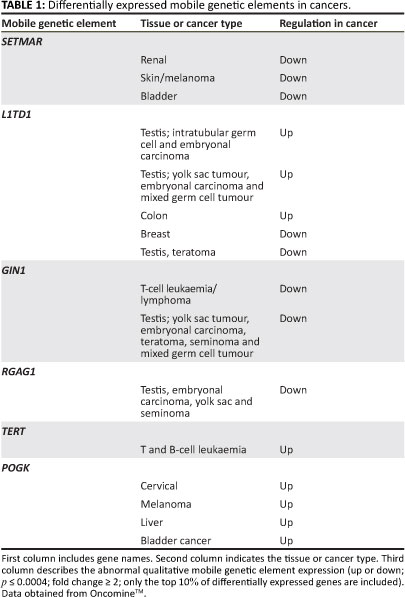
To investigate whether the disruption of MGE behaviour is a feature of most cancer cell types, we analysed the expression profiles of MGE terms (N=26, supporting information S1) indentified in the Affymetrix annotation portal (Netaffx) across diverse cancer cell populations and compared these with non-cancer cells for the same tissue type using the OncomineÂTM online (http://www.oncomine. org/) repository of cancer microarray expression.41 The OncomineTM repository consists of published microarray data sets of a diverse assortment of cancers. For this analysis only tumour versus healthy tissue comparisons were used. These comparisons included various matched controls, that is, non-cancer cells from the same tissue in the same individual and non-cancer cells from the same tissue but different individuals (for example in the case of leukaemia where healthy blood or bone marrow cannot be obtained). MGE terms included transposons, non-LTR and LTR retrotransposons (e.g. LINE 1), MGE regulatory genes (e.g. SUPT5H), as well as partially domesticated genes with MGE domains such as SETMAR and POGK, which may be selfish or cooperative in their behaviour.42 The OncomineÂTM database was queried using the derived MGE gene list and examined for expression differences. Data sets were filtered with the default OncomineÂTM criteria, namely p <0.0004, fold change > 2 and the 10th percentile.
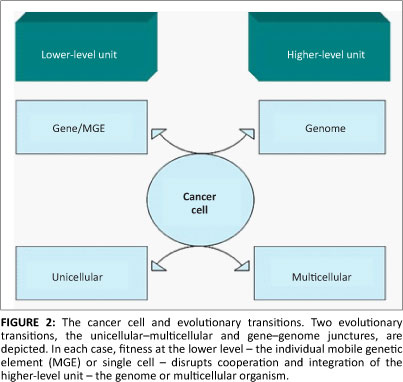
Data from the OncomineTM database revealed altered MGE expression in a diverse array of malignant tissues (Table 1 and Figure 1). Particularly prevalent were germ-cell tumours (p<0.0004) where some MGEs had either lower or higher expression. For example, the retrotransposon L1TDT1 was differentially expressed in two studies of germ-cell and embryonal cancers.43,44 Differential expression of MGE domains (such as SETMAR and POGK) was common in epithelial tumours, particularly in bladder and skin tumours. These observations support the hypothesis that there is differential expression of various MGEs in a diverse range of cancer cell types. There is a change in the reproduction rate which implies a change in life-history strategy in the cancer cell compared with the non-cancer cell. The altered MGE life-history strategy reflects a difference in the gene and genome levels of selection in cancer cells compared with non-cancer cells. Such changes in life-history strategy typically, but not always, reflect an increase in competition between lower-level units or increased tension between lower and higher levels of complexity.24,30 Interestingly, some patterns emerge with regard to MGE expression and tumour types. For example, germ-line tumours such as the testicular cancers are represented more frequently. Whether these observations have any evolutionary significance is not clear and would require more detailed investigations. It was noted that the expression of partially domesticated or host-MGE hybrids like SETMAR (Table 1) and selfish, non-domesticated MGEs like the LINE 1 domain L1TD1 were both affected. This observation suggests that the transformation to malignancy impacted the behaviour of MGE domains irrespective of their level of domestication. The data reported here, however, cannot distinguish between gene or genome level activity of the hybrid or partially domesticated MGE domains like SETMAR and POGK (for a discussion of partially domesticated MGEs and evolutionary transitions see Durand and Michod28 and Sinzelle et al.42). Some MGE domains such as GIN1, RGAG1 and RGAG4 demonstrated lower expression in some cancers when compared with controls, indicating that their change in life-history strategy may lead to up or down regulation. Overall, these data revealed that aberrant expression of MGE domains was a feature of the majority of cancers examined and that both gene-genome and unicellular-multicellular junctures are impacted in the cancer cell (Figure 2).
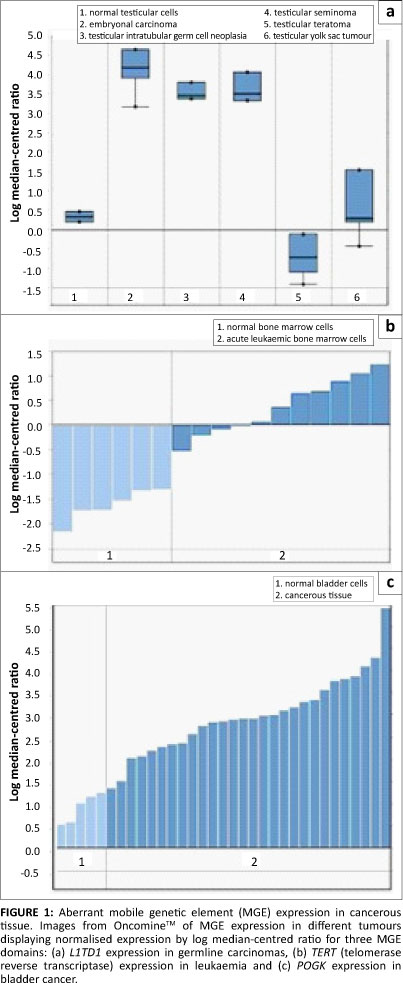
Not all cancers, however, reflect the tension between genes and genomes. Some cancers, such as chronic myeloid leukaemia, can be defined by specific gene mutations. In chronic myeloid leukaemia, chromosomal translocation of chromosomes 9 and 22 gives rise to a fused gene product (BCR-ABL1) which provides mutated cells with a proliferative advantage. In this case, the mutant chimera will increase its frequency within a population of haematopoietic cells over wild-type bcr and abl1. However, this mutation provides a benefit (albeit a short-term one) to its nearest higher level, that is, selection of the mutant BCR-ABL1 gene will select for genomes (cells) that carry this mutant. In this situation, there is initially no conflict between the gene and the genome. For selection to shift to the gene level, the gene must become an individual in its own right, just as a transformed cell develops a life history independently of the organism. Additionally, this newly acquired individuality is frequently harmful to the higher level. Changes to gene-level life-history strategies may indeed occur in some cancers (for example see Deming et al.45). Some MGEs are truly 'selfish' elements. Their reproduction within the genome is deleterious to the rest of the host genome34 just like any other host-parasite relationship. In the case of MGE fitness, overexpression increases reproduction and underexpression increases viability.28 But why and how does the cancer cell mirror these ancient evolutionary transitions?
Evolutionary transitions and conflict
For the transition to a higher level of biological complexity to occur, lower-level conflicts need to be controlled such that selection at the higher level is maintained. This argument is now well established in evolutionary transition theory,17 particularly in the context of multicellularity origins20 and is equally applicable to the gene-genome transition.28 There is always the temptation for lower-level units to cheat or not cooperate, thereby disrupting the emergence or maintenance of a higher-level individual. Lower-level units might, for example, replicate faster and increase their representation in the next generation at a cost to the higher level. A number of the features of evolutionary transitions mean that disintegration of the higher level into its lower-level units can be avoided. The division of labour, for example, means that organisation at the higher level is functionally integrated and lower-level units are no longer capable of independent replication. This characteristic of contingent irreversibility17 ensures cooperation between lower-level units, which are not autonomous and have no independent future. The temptation to cheat and develop an independent existence, however, always exists and mechanisms for controlling lower-level conflicts and maintaining higher-level functionality have emerged to counteract this tendency.
Controlling mechanisms exist that maintain both genome integrity and multicellularity. With respect to genome integrity, genoprotective mechanisms are essential for regulating MGE behaviour and genome maintenance30 and are ubiquitous in nature. A disruption of these gene-level policing mechanisms means that MGEs are no longer controlled and are free to proliferate or indulge in selfish behaviour. Cancer genomes undergo numerous and large-scale changes and dysfunctional genoprotective mechanisms are almost certain to occur during cancer progression. It is no surprise, therefore, to find that a disruption of the gene-genome transition occurs in cancer cells. Similarly, in multicellular organisms the immune system polices the activity and reproduction of individual cells. However, mutant genomes that allow cells to escape immune surveillance are free to indulge in selfish reproduction and regress to their lower-level unicellular-like existence. It is interesting that while cancers incur a fitness cost to the higher level multicellular organism, some tumours have regressed to a truly unicellular existence. These tumours include the peculiar cases of canine venereal transmissible carcinoma21 and a facial tumour in Tasmanian devils.22,46 In these cases, the cancer cells are genetically unrelated to their hosts, but are transmitted between individuals through physical contact. In summary, it is the failure of mechanisms that control lower-level conflict that allows the cancer cell to mirror the ancient evolutionary transitions discussed here.
Other levels of complexity in malignant cells
The above data and arguments indicate that two major evolutionary transitions are disrupted in cancer cells. Is it reasonable to suspect that other levels of complexity are found in malignancy? A superficial consideration of events known to take place in tumours, suggests the answer is yes. Although the criteria for what may constitute a major or minor evolutionary transition can be debated,14 other conflicts between evolutionary units occur in cancer cell populations. For example, competition and conflict between chromosomes for increased representation in the next generation is known to occur.47,48 Whether the variation in chromosome number (aneuploidy) is a cause or consequence of cancer is again a vexed question, but as with MGEs in cancer, the point here is that conflict arises and lower-level units invest more in their own fitness. Similarly, intergenomic conflict, such as that which may occur between mitochondrial and nuclear genomes,47,49 may cause variations in mitochondrial gene or genome number in cancer cells. These intragenomic and intergenomic conflicts suggest that other tensions between levels of complexity may be playing out.
Concluding remarks
According to MLST, multiple levels of complexity may act as levels of selection. For these increases in complexity to have formed, cooperation was required. Malignancy represents the failure of cooperation and the subsequent increases in tension between lower and higher levels of complexity. This paper highlights cooperation and conflict in cancer occurring between two evolutionary transitions: the unicellular-multicellular and the gene-genome.
Investigating carcinogenesis from an evolutionary perspective provides conceptual advances, enhances our understanding of the pathological process and may lead to new treatment strategies. It is conceivable that therapeutic interventions that account for the evolutionary processes inherent in cancer cells are more likely to succeed. For example, therapeutic interventions that intentionally (or as a side effect) promote the disintegration of the evolutionary transitions discussed above may actually exacerbate the disease, whereas others which promote cooperation diminish it.
Acknowledgements
J.F. is supported by the Department of Molecular Medicine and Haematology, University of the Witwatersrand and a National Research Foundation Bioinformatics and Functional Genomics Programme grant. P.M.D. is supported by the University of the Witwatersrand Faculty of Health Sciences Individual Research Grant and IBM.
Competing interests
We declare that we have no financial or personal relationships which may have inappropriately influenced us in writing this paper.
Authors' contributions
J.F. performed the experimental work. J.F. and P.D. conceptualised the project, analysed the data and wrote the manuscript.
References
1. Gatenby RA, Vincent TL. An evolutionary model of carcinogenesis. Cancer Res. 2003;63(19):6212-220. [ Links ]
2. Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924-935. http://dx.doi.org/10.1038/nrc2013 [ Links ]
3. Pepper JW, Scott Findlay C, Kassen R, Spencer SL, Maley CC. Cancer research meets evolutionary biology. Evol Appl. 2009;2:62-70. http://dx.doi.org/10.1111/j.1752-4571.2008.00063.x [ Links ]
4. Crespi B, Summers K. Evolutionary biology of cancer. Trends Ecol Evol. 2005;20(10):545-552. http://dx.doi.org/10.1016/j.tree.2005.07.007 [ Links ]
5. Nagy JD. Competition and natural selection in a mathematical model of cancer. Bull Math Biol. 2004;66(4):663-687. http://dx.doi.org/10.1016/j.bulm.2003.10.001 [ Links ]
6. Heng HH, Stevens JB, Bremer SW, Liu G, Abdallah BY, Ye CJ. Evolutionary mechanisms and diversity in cancer. Adv Cancer Res. 2011;112:217-253. http://dx.doi.org/10.1016/B978-0-12-387688-1.00008-9 [ Links ]
7. Heng HH. The genome-centric concept: Resynthesis of evolutionary theory. Bioessays. 2009;31(5):512-525. http://dx.doi.org/10.1002/bies.200800182 [ Links ]
8. Heng HH, Liu G, Stevens JB, et al. Decoding the genome beyond sequencing: The new phase of genomic research. Genomics. 2011;98(4):242-252. http://dx.doi.org/10.1016/j.ygeno.2011.05.008 [ Links ]
9. Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390(6658):404-407. http://dx.doi.org/10.1038/37126 [ Links ]
10. Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA. 2008;105(11):4283-4288. http://dx.doi.org/10.1073/pnas.0712345105 [ Links ]
11. Domazet-Loso T, Tautz D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010;8:66. http://dx.doi.org/10.1186/1741-7007-8-66 [ Links ]
12. Hauptmann S, Schmitt WD. Transposable elements - is there a link between evolution and cancer? Med Hypotheses. 2006;66(3):580-591. http://dx.doi.org/10.1016/j.mehy.2005.08.051 [ Links ]
13. Goodnight CJ, Stevens L. Experimental studies of group selection: What do they tell us about group selection in nature? Am Nat. 1997;150 Suppl 1:S59-S79. http://dx.doi.org/10.1086/286050 [ Links ]
14. Griesemer J. The units of evolutionary transition. Selection. 2000;1:67-80. [ Links ]
15. Okasha S. Evolution and the levels of selection. Oxford: Clarendon Press; 2006. http://dx.doi.org/10.1093/acprof:oso/9780199267972.001.0001 [ Links ]
16. Thompson NS. Shifting the natural selection metaphor to the group level. Behav Philos. 2000;28:83-101. [ Links ]
17. Szathmary E, Smith JM. The major evolutionary transitions. Nature. 1995;374(6519):227-232. http://dx.doi.org/10.1038/374227a0 [ Links ]
18. Michod RE. Darwinian dynamics: Evolutionary transitions in fitness and individuality. Princeton: Princeton University Press; 1999. [ Links ]
19. Michod RE. Evolution of individuality during the transition from unicellular to multicellular life. Proc Natl Acad Sci USA. 2007;104 Suppl 1:8613-8618. http://dx.doi.org/10.1073/pnas.0701489104 [ Links ]
20. Buss LW. The evolution of individuality. Princeton: Princeton University Press; 1987. [ Links ]
21. Murgia C, Pritchard JK, Kim SY, Fassati A, Weiss RA. Clonal origin and evolution of a transmissible cancer. Cell. 2006;126(3):477-487. http://dx.doi.org/10.1016/j.cell.2006.05.051 [ Links ]
22. Siddle HV, Kreiss A, Eldridge MD, et al. Transmission of a fatal clonal tumor by biting occurs due to depleted MHC diversity in a threatened carnivorous marsupial. Proc Natl Acad Sci USA. 2007;104(41):16221-16226. http://dx.doi.org/10.1073/pnas.0704580104 [ Links ]
23. Michod RE. On the transfer of fitness from the cell to the multicellular organism. Biol Philos. 2005;20:967-987. http://dx.doi.org/10.1007/s10539-005-9018-2 [ Links ]
24. Michod RE, Nedelcu AM. On the reorganization of fitness during evolutionary transitions in Individuality. Integr Comp Biol. 2003;43:64-73. http://dx.doi.org/10.1093/icb/43.1.64 [ Links ]
25. Durand P, Coetzer T. Evolutionary biology and drug development. In: Kapetanovic IM, editor. Drug discovery and development - present and future. Rijeka: Intech Books, 2011; pp. 24-42. [ Links ]
26. Weinberg R. The biology of cancer. Oxford: Garland Science; 2008. [ Links ]
27. Domazet-Loso T, Brajkovic J, Tautz D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. [ Links ]
Trends Genet. 2007;23(11):533-539. http://dx.doi.org/10.1016/j.tig.2007.08.014
28. Durand PM, Michod RE. Genomics in the light of evolutionary transitions. Evolution. 2010;64:1533-1540. http://dx.doi.org/10.1111/j.1558-5646.2009.00907.x
29. Brosius J. Genomes were forged by massive bombardments with retroelements and retrosequences. Genetica. 1999;107(1-3):209-238. http://dx.doi.org/10.1023/A:1004018519722 [ Links ]
30. Kidwell MG, Lisch DR. Perspective: Transposable elements, parasitic DNA, and genome evolution. Evolution. 2001;55(1):1-24. [ Links ]
31. Le Rouzic A, Capy P. The first steps of transposable elements invasion: Parasitic strategy vs. genetic drift. Genetics. 2005;169(2):1033-1043. http://dx.doi.org/10.1534/genetics.104.031211 [ Links ]
32. Romanish MT, Cohen CJ, Mager DL. Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin Cancer Biol. 2010;20(4):246-253. http://dx.doi.org/10.1016/j.semcancer.2010.05.005 [ Links ]
33. Belancio VP, Roy-Engel AM, Deininger PL. All y'all need to know 'bout retroelements in cancer. Semin Cancer Biol. 2010;20(4):200-210. http://dx.doi.org/10.1016/j.semcancer.2010.06.001 [ Links ]
34. Kim DS, Huh JW, Kim HS. Transposable elements in human cancers by genome-wide EST alignment. Genes Genet Syst. 2007;82(2):145-156. http://dx.doi.org/10.1266/ggs.82.145 [ Links ]
35. Lamprecht B, Walter K, Kreher S, et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med. 2010;16(5):571-579. http://dx.doi.org/10.1038/nm.2129 [ Links ]
36. Miki Y, Nishisho I, Horii A, et al. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992;52(3):643-645. [ Links ]
37. Morse B, Rotherg PG, South VJ, Spandorfer JM, Astrin SM. Insertional mutagenesis of the myc locus by a LINE-1 sequence in a human breast carcinoma. Nature. 1988;333(6168):87-90. http://dx.doi.org/10.1038/333087a0 [ Links ]
38. Stribinskis V, Ramos KS. Activation of human long interspersed nuclear element 1 retrotransposition by benzo(a)pyrene, an ubiquitous environmental carcinogen. Cancer Res. 2006;66(5):26l6-2620. http://dx.doi.org/10.1158/0008-5472.CAN-05-3478 [ Links ]
39. Ting DT, Lipson D, Paul S, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331(6017):593-596. http://dx.doi.org/10.1126/science.1200801 [ Links ]
40. Wurmbach E, Chen YB, Khitrov G, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45(4):938-947. http://dx.doi.org/10.1002/hep.21622 [ Links ]
41. Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1-6. [ Links ]
42. Sinzelle L, Izsvak Z, Ivics Z. Molecular domestication of transposable elements: From detrimental parasites to useful host genes. Cell Mol Life Sci. 2009;66(6):1073-1093. http://dx.doi.org/10.1007/s00018-009-8376-3 [ Links ]
43. Korkola JE, Houldsworth J, Chadalavada RS, et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66(2):820-827. http://dx.doi.org/10.1158/0008-5472.CAN-05-2445 [ Links ]
44. Skotheim RI, Lind GE, Monni O, et al. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005;65(13):5588-5598. http://dx.doi.org/10.1158/0008-5472.CAN-05-0153 [ Links ]
45. Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification in breast cancer: A meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000;83(12):1688-1695. http://dx.doi.org/10.1054/bjoc.2000.1522 [ Links ]
46. Murchison EP, Tovar C, Hsu A, et al. The Tasmanian devil transcriptome reveals Schwann cell origins of a clonally transmissible cancer. Science. 2009;327(5961):84-87. http://dx.doi.org/10.1126/science.1180616 [ Links ]
47. Burt A, Trivers R. Genes in conflict: The biology of selfish genetic elements. Boston: Harvard University Press; 2006. [ Links ]
48. Pellman D. Cell biology: Aneuploidy and cancer. Nature. 2007;446(7131):38-39. http://dx.doi.org/10.1038/446038a [ Links ]
49. Rand DM. Mitochondrial genetics of aging: Intergenomic conflict resolution. Sci Aging Knowledge Environ. 2005;2005(45):re5. http://dx.doi.org/10.1126/sageke.2005.45.re5 [ Links ]
 Correspondence to:
Correspondence to:
Jonathan Featherston
Department of Molecular Medicine and Haematology, University of the Witwatersrand Medical School, 7 York Road, Parktown 2193, South Africa
Email: jonathan.featherston@nhls.ac.za
Received: 09 Nov. 2011
Accepted: 08 May 2012
Published: 04 Sept. 2012
© 2012. The Authors. Licensee: AOSIS OpenJournals. This work is licensed under the Creative Commons Attribution License.

