










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.108 n.3-4 Pretoria Jan. 2012
RESEARCH ARTICLES
Detection and molecular identification protocols for Phyllosticta citricarpa from citrus matter
Linda MeyerI; René JacobsI; Jan M. KotzéII; Mariette TruterIII; Lise KorstenI
IDepartment of Microbiology and Plant Pathology, University of Pretoria, Pretoria, South Africa
IIConsultant: Citrus Industry, Kokanje, Modimolle, South Africa
IIIBiosystematics, ARC-Plant Protection Research Institute, Pretoria, South Africa
ABSTRACT
Strict quarantine measures for the export of South African citrus fruit to European and US markets require the development of sensitive and accurate detection methods for the pathogen Phyllosticta citricarpa - a fungus causing citrus black spot disease. Because of the presence of other, non-pathogenic Phyllosticta species, rapid and accurate verification of the Phyllosticta species present on exported citrus fruit is important to producers, exporters and regulatory authorities to prevent unnecessary losses. We have analysed over 800 samples collected over 7 years and have compared sample preparation and detection protocols applied in different environments: nurseries, production systems including phytosanitary inspections in orchards, pack houses and export terminals in order to compile protocols for the detection of P. citricarpa. Standard procedures of sample preparation and DNA extraction were adapted to suit diverse inoculum sources. Low pathogen numbers in symptomless green leaves, for example, obliged the use of a wet-dry enrichment technique constituting the stimulation of fungal growth for easier detection. Physical maceration was adapted for sturdy material using liquid nitrogen or bead beating. The use of a two-step polymerase chain reaction (PCR) with nested primers significantly increased both the sensitivity and the specificity of the PCR performed on soil samples, overcoming problems with relatively impure DNA extracts and low pathogen numbers. The assays have proven to be highly consistent, thereby providing a reliable, reproducible and highly sensitive detection and diagnostic service to the southern African citrus industries in order to sustain market access.
Introduction
Citrus black spot (CBS) is a disease caused by the ascomycetous fungus Guignardia citricarpa Kiely (the anamorph or asexual stage is termed Phyllosticta citricarpa [McAlpine] Aa). Presently, CBS is widespread within some of the major citrus producing countries of the world, such as Australia, Argentina, Brazil, India and South Africa. The disease has never been recorded in the United States of America, Chile, New Zealand, Israel or any European country.1 Stern regulatory measures that restrict market access for countries with CBS to CBS-free markets have necessitated the development of sensitive and accurate detection methods for P. citricarpa.2,3,4
The accurate detection of P. citricarpa and the disease it causes has been complicated by the presence of several other, non-pathogenic, species in the genus.3,5,6,7,8,9 Of these fungi, the best known is the common endophyte Phyllosticta capitalensis Henn. that occurs on many tropical and subtropical native and crop plants.5,10,11 Lesions on citrus fruit colonised by P. capitalensis, tested using earlier protocols such as the incubation of infected material, microscopic examination and dissecting and plating of lesion pieces, have been misdiagnosed as CBS, resulting in significant financial loss to exporters.42,13 Recently, two additional Phyllosticta species, Phyllosticta citriasiana Wulandari, Crous & Gruyter and Phyllosticta citribrazilliensis C. Glienke & Crous, were isolated from necrotic spots on pomelo fruit in Asia14 and Brazil,5 respectively. Apart from the difficulty in accurately distinguishing these species of Phyllosticta by means of morphological characteristics, the process is also time consuming (taking between 7 and 10 days). Rapid and accurate verification of the Phyllosticta species present on exported fruit is of the utmost importance to the producer, exporter and regulatory authority.
A range of articles has been published on the use of highly specific and rapid polymerase chain reaction (PCR) techniques to detect and distinguish between P. citricarpa and P. capitalensis. A variety of primer sets has been developed for P. citricarpa and P. capitalensis by different research groups.2,3,4,15,16 Peres and others3 have evaluated and compared these various molecular systems, with emphasis on DNA extraction techniques and primer sensitivity. Although they concluded that most of the primer sets evaluated were species-specific and useful for detection, they had difficulty in extracting sufficient, quality DNA from single fruit spots, mentioning low sensitivity and time consuming, expensive procedures. Van Gent-Pelzer et al.15 went one step further and developed an effective and sensitive real-time PCR system for P. citricarpa, but still had difficulty in extracting DNA from single fruit spots. With both standard PCR and real-time PCR, and irrespective of which primer set is used, focused sample or plant tissue preparation and DNA extraction are essential for successful detection of the black spot pathogen in plant material. Often a single fruit spot is all that is available for pathogen detection.
The development and application of sampling and DNA extraction protocols include adaptations to standard procedures to suit various inoculum sources. The low pathogen numbers in symptomless green leaves, for example, obliges an enrichment step constituting the stimulation of fungal growth for easier detection.12,17,18,19 Soil is considered to be a complex environment and working with DNA recovered from soil is often problematic because of the presence of PCR-inhibiting chemical components in soil.20,21 Therefore, PCR-based studies require extensive DNA extraction methods and careful purification of nucleic acid extracts in order to remove humic acids and other contaminants. DNA losses during extensive purification may compromise the detection of pathogens at low concentrations.22,23 This possibility, therefore, compels the adaptation of techniques to sensitise detection. The use of a two-step PCR with nested primers increases both the sensitivity and the specificity of the PCR significantly, which can overcome problems with relatively impure DNA extracts and low pathogen numbers.
In this paper, we describe the different sampling protocols and methods developed, validated and extensively used to detect P. citricarpa and distinguish it from P. capitalensis, the only other Phyllosticta species known to occur on citrus in South Africa. These methods include detection from cultures, symptomless and symptomatic leaves, fruit, twigs, petioles, soil and spore traps.
Materials and methods
Sample preparation and DNA extraction
The cultures and plant material that were used in this study are summarised in Table 1.
Isolation from clean or mixed fungal cultures using the DNeasyTM extraction technique
Cultures were grown on different fungal growth media, namely, potato dextrose agar, malt extract agar and oats agar. Using a clean, flamed scalpel, a section of fungal growth was removed from pure or mixed culture plates. Fungal growth typical for Phyllosticta was randomly selected taking special care to limit the use of pigmented mycelium. The material was placed in microcentrifuge tubes containing sterile zirconium or silica beads of different sizes and macerated, together with extraction buffer (DNeasy® Plant Mini kit, Qiagen, Cape Town, South Africa), by a bead beater instrument (FastPrep® Instrument, Qbiogene Inc., Montreal, Canada) for 15 s at 4 m/s. DNA extraction was continued using the manufacturer's standard protocol for DNA isolation.24
Isolation from clean or mixed fungal cultures using Whatman FTATM technology
A section of fungal growth was aseptically removed from a culture as described for the DNeasy extraction technique, but oozing pycnidiospores (when present) were preferentially selected. The mycelium and/or spores were smeared onto a FTATM matrix card (Whatman, Maidstone, UK) and extraction was performed using the standard protocol.25
Isolation from green, wilted or dry citrus leaves
Symptomless green leaves were treated with a 'wet-dry' technique to enrich fungal mycelial mass and stimulate fruiting body formation. The technique included alternate daily wetting and drying of leaves. Leaves were rinsed in tap water to remove excess dirt, after which surfaces were disinfected with sodium hypochlorite (1.5% NaOC1) for 2 min, followed by thorough rinsing with sterile water. The following four steps were repeated for 4-10 consecutive days: (1) leaves were submerged in sterile tap water at 35 °C for 30 min, (2) excess water was removed by draining the leaves on paper towels for 5 min, (3) leaves were placed in plastic bags (250 mm x 380 mm x 20 μm) and incubated at 42 °C for 6 h and (4) the leaves were air dried at room temperature (22 °C - 26 °C) for 17.5 h under fluorescent light in open bags. After 4 days, leaf material with noticeable mycelium colonisation underwent PCR. Optimally wilted leaves were paper brown and leathery with minimal saprophytic fungal growth. By continuing the process, fruiting bodies (mostly pycnidia) were noticed after about 8 days. After 10 days the fruiting bodies were well defined and, depending on the CBS incidence and the success of colonisation, were abundant.
In the case of symptomatic green leaves and leaf litter with fruiting bodies, leaves were used directly for DNA extraction without the wet-dry treatment. A selection of 5 to 12 leaves was made per sample, giving preference to leaves with typical black spot lesions, visible pseudothecia or with darker coloured areas. When no pseudothecia or fungal structures were visible on leaf litter, leaves were selected randomly and no more than two punches (2 mm, Unicore punch, Whatman) were removed from each leaf. A maximum of 10-12 leaf pieces was removed from the selected leaves with a 2-mm Unicore punch (Whatman). The plant tissue was placed into a microcentrifuge tube containing extraction buffer AP1, RNase and two sterile 6.35-mm ceramic beads (Qbiogene, Montreal, Canada). The material was ground in a bead beater instrument for 25 s at 5 m/s and DNA was immediately extracted using the Qiagen DNeasy Plant Mini kit standard protocol for DNA isolation.24
Isolation from symptomatic or symptomless fruit
Fruit were surface sterilised with sodium hypochlorite (1.5% NaOC1) for 2 min, followed by thorough rinsing in sterile water. A thin layer of the fruit flavedo containing the black spot lesion and the surrounding tissue was removed with a sharp, sterilised scalpel. Exposed tissue was cut into 2 mm x 2 mm squares, removed and placed into 1.5-mL microcentrifuge tubes. If the lesion was too small, the surface layer of the flavedo was not removed. A maximum of two small (1 mm - 2 mm diameter) lesions per extraction was used. Symptomless fruit were treated the same, except four small sections (1 mm - 2 mm diameter) were randomly cut from the surface. The closed tube was submerged into liquid nitrogen with forceps for 20 s - 30 s and the plant material was then ground manually with a sterile microhomogeniser and the step was repeated twice. The AP1 extraction buffer (DNeasy® Plant Mini DNA extraction kit, Qiagen) was added before the tissue thawed, and the sample was kept on ice until all samples were prepared. DNA extraction was continued using the manufacturer's standard protocol.24
Isolation from twigs and petioles
A 1-mm thick layer of the twig or petiole surface containing lesions and/or fruiting bodies was removed. A maximum of six small (1 mm - 2 mm diameter) lesions per extraction were used. The tissue was cut into smaller pieces and placed into microcentrifuge tubes containing extraction buffer, RNase and two ceramic beads (6.35 mm, Qbiogene). Material was macerated in a bead beater for 30 s at 5.5 m/s. Buffer volumes were adjusted to account for the volume of the beads and the Qiagen DNeasy standard protocol for DNA isolation was used.24
Isolation from soil
Soil from the top layer of the A horizon of a citrus tree rhizozone was collected after clearing infected leaf litter.
The soil was added to a microcentrifuge tube containing zirconium or silica beads that were either 0.5 mm or 1 mm in diameter (Biospec Products, Bartlesville, OK, USA). Soil DNA extraction buffer and proteinase K were added and the tube was agitated with a bead beater for 20 s at 5 m/s. Soil lysis buffer was added and the standard protocol and volumes described in the SoilMasterTM DNA extraction kit manual (Epicentre Biotechnologies, Madison, WI, USA) were further applied.26
Isolation from microscope slide from Kotzé Inoculum Monitor
Standard microscope slides were coated with petroleum jelly and used in a Kotzé Inoculum Monitor (Interlock Systems, Pretoria, South Africa) to capture spores from citrus leaf litter.27 The presence of Guignardia ascospores on the slide was microscopically verified without using any stain or mounting fluid. Using a clean, flamed scalpel, a thin layer of petroleum jelly was scraped off and smeared onto a FTA matrix card and left to dry completely. The standard Whatman FTATM DNA extraction protocol was used.25
Polymerase chain reaction amplification
Standard polymerase chain reaction conditions
For detection and differentiation of P. citricarpa and P. capitalensis, the primers CITRIC1 and CAMEL2 were used together with the ITS4 reverse primer.4 PCR reactions were performed in 50-μL volumes, with each reaction containing 2 μL template DNA, 15 pmol ITS4, 10 pmol CITRIC1 and 60 pmol CAMEL2, 5 μL recommended 10x buffer (supplied with Taq polymerase), 200 μM dCTP, dGTP, dATP and dTTP (TaKaRa Bio Inc., Shiga, Japan) and 0.5 U Taq polymerase (TaKaRa). Following an initial denaturation step at 95 °C for 2 min, 35 PCR cycles were performed on an Eppendorf G thermocycler (Eppendorf, Hamburg, Germany) using the following conditions: a denaturation step at 94 °C for 30 s followed by annealing at 58 °C for 45 s and extension at 72 °C for 90 s. These cycles were followed by a final extension at 72 °C for 7 min. The amplified DNA fragments were visualised on a 1.5% (w/v) agarose gel in tris-borate-EDTA (TBE) buffer.28 Purified positively identified DNA extracts from P. citricarpa and P. capitalensis were included as positive controls. A citrus isolate of Colletotrichum gloeosporioides (Penz.) Penz. & Sacc. (because of its ubiquitous occurrence on citrus) and distilled water were included as negative controls in all PCR reactions.
Nested polymerase chain reaction conditions
A two-step PCR was developed using the universal primers ITS1 and ITS4 to amplify the internal transcribed spacer (ITS) region of any fungal tissue present in the sample. Amplicons of the first PCR step were used for a second amplification with primers CITRIC1 and CITRIC1R. Therefore, the second PCR used the product of the first PCR as template and a second, inner pair of primers nested within the region amplified by the first PCR.
The design of the oligonucleotide reverse primer CITRIC1R (5'-GAA AGG TGA TGG AAG GGA G-3') was based on the DNA sequences of previously sequenced ITS gene regions of Phyllosticta species10 and was used in conjunction with the primer CITRIC1. In order to confirm the specificity of the primer, it was analysed using the Basic Local Alignment Search Tool (BLAST) program29 (National Centre for Biotechnology Information). Previously sequenced isolates5 were used to evaluate the efficacy of the CITRIC1-CITRIC1R primer set. Standard PCR analyses were conducted on these isolates as described above.
PCR reactions were performed in 25-μL volumes, with each reaction containing 1 μL template DNA, 20 pmol CITRIC1, 20 pmol CITRIC1R, 2.5 μL recommended 10x buffer (TaKaRa), 200 μM dNTP (TaKaRa) and 0.3 U Taq polymerase (TaKaRa). Following an initial denaturation step at 95 °C for 2 min, 40 PCR cycles were performed on an Eppendorf G thermocycler using the following conditions: a denaturation step at 94 °C for 30 s followed by annealing at 60 °C for 45 s and extension at 72 °C for 90 s, followed by a final extension at 72 °C for 7 min. The amplified DNA fragments were visualised on a 1.5% (w/v) agarose gel in TBE buffer.28 The same positive and negative controls were used as described for the standard PCR conditions.
Results and discussion
Recent developments in molecular biology have helped to alleviate many of the challenges associated with the movement, control and regulation of quarantine pests. However, these molecular techniques are not always universally accepted, because of the need to validate the techniques and determine limitations, specifically with regard to specificity.30,31 In this paper, we have described optimised and validated techniques for the successful DNA extraction of P. citricarpa from various citrus samples, to ultimately provide good quality and sufficient DNA for downstream PCR tests and trials. The extraction protocols described in the current study form part of a test method that is used in ISO/IEC 17025 accredited laboratories. This accreditation implies that the method has been evaluated extensively and validated by our laboratory as well as outside laboratories as part of a mandatory interlaboratory testing scheme. In all cases, the assays were shown to be reliable, reproducible and highly sensitive. In this study, both standard and nested PCRs were performed on 823 samples and were used to successfully identify P. citricarpa from 529 samples (Table 1).
The extensive use of this PCR-based technique with Phyllosticta specific primers for diagnosis and differentiation of P. citricarpa and P. capitalensis has proven to be consistently repeatable and reliable. A high concentration of pure, undamaged, DNA could be successfully and consistently extracted from cultures (139 samples), leaves (222 samples), fruit (245 samples), twigs (20 samples), petioles (10 samples) and soil (125 samples) using commercially available extraction kits in combination with bead beating. The bead beater served to physically disrupt plant and fungal tissue. The use of different beads in combination with different durations and force (m/s) of beating was optimised in such a way that DNA was not damaged but plant and fungal cells were disrupted. For sturdy material such as leaves, twigs and petioles, two 6.35-mm ceramic beads were ideal and could disrupt the tissue in a very short time, with minimal DNA damage. In contrast, fungal mycelium and spores required only a mix of small zirconium beads to break the fungal cell walls. The addition of a physical disruption step to the DNA extraction protocol greatly increased the quality and quantity of the DNA extracted.
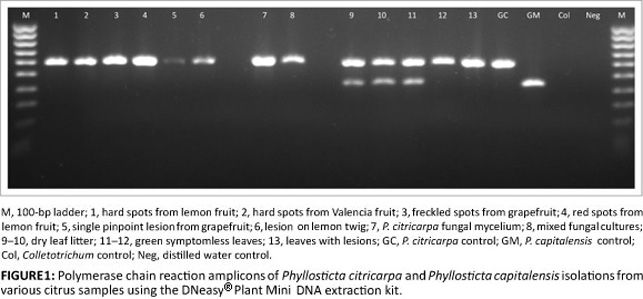
For isolation from infected or symptomless fruit, a very small amount of lesion tissue, regardless of the symptom type, was required to produce a sufficient DNA concentration for PCR detection. Usually, a maximum of two lesions (1 mm - 2 mm diameter) was used for successful extraction and amplification (Figure 1). In samples where larger host tissue sections were included, organic inhibitors in the host tissue brought about false negative results. Often fruit containing questionable spots had only one or two lesions from which to extract DNA, making it imperative that no material was lost. By using liquid nitrogen with a microhomogeniser when working with limited tissue samples, no material was lost and DNA was protected for optimal extraction. A maximum of two or three lesions from twigs and petioles could also be homogenised in liquid nitrogen, but two 6.35-mm ceramic beads used with the bead beater were just as effective in disrupting the tissue and required less time (Figure 1).
Positive results were consistently achieved with the detection of Phyllosticta species from symptomless green leaves using the empirical wet-dry technique (Figure 2). Phyllosticta species can survive endophytically in green citrus leaves, but in small localised zones only. The wet-dry technique (similar to natural leaf wilting) stimulated the change to saprophytic growth at an accelerated rate. This acceleration enhanced the likelihood of detecting single infection points already present in citrus nursery trees, ultimately curbing the spread of this quarantine pathogen to new orchards and growing areas.
FTA matrix cards were designed for the collection, archiving and purification of nucleic acids from a wide variety of biological samples for PCR analysis. The great advantage of the FTA technology is that the samples can be dried and stored for years. Isolation from clean or mixed fungal cultures (54 samples) using FTA matrix technology, in contrast with the DNeasy extraction technique, produced a lower concentration of DNA, albeit sufficient to yield a positive PCR. FTA matrix technology was also effectively used in the isolation of DNA from petroleum-covered microscope slides retrieved from the inoculum monitor (28 samples). The absence or presence of pathogenic ascospores was thus confirmed. Nevertheless, the fluctuating release of spores from leaf litter should be considered.
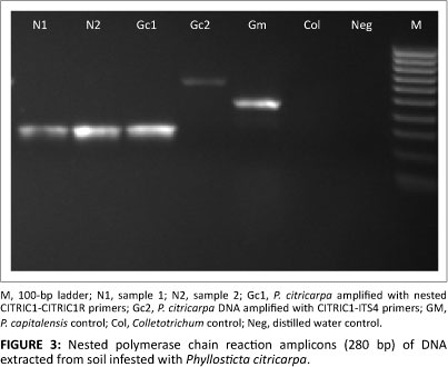
Isolation from soil proved to be effective with the use of the Soil MasterTM kit (Epicentre). The direct PCR system often failed to amplify the fragment of interest from the total DNA extracted from natural soil underneath infected leaf litter. Where amplicons could be obtained, they were often rather faint. Therefore a nested PCR was developed based on an initial amplification using ITS1 and ITS4 primers, followed by a subsequent amplification with CITRIC1-CITRIC1R primers. A distinct ITS fragment of 280 bp could be reproducibly generated for 87 soil samples (Figure 3). Nested PCR was more sensitive in situations where inhibitory substances or low DNA concentrations prevailed.
The protocols we have described here have been extensively used to confirm and clarify the presence or absence of the pathogen in questionable fruit spots for the producer, pack house manager, export agent, quarantine and quality control officials, and researchers. Pathogen detection in soil, twig, petiole and leaf samples continuously helps to paint a clearer picture of the spread and survival of the pathogen P. citricarpa and its common endophytic partner P. capitalensis in South Africa. The protocols focusing on pathogen isolation from symptomless green leaves have been successfully implemented by the citrus industry in South Africa to screen citrus nurseries for the presence of P. citricarpa, to improve quality and hygiene and to prevent the spread of black spot to new orchards.
Acknowledgements
The financial support of Citrus Research International (CRI) and the Technology and Human Resources for Industry Programme (THRIP), a partnership programme funded by the Department of Trade and Industry and managed by the National Research Foundation, is gratefully acknowledged.
Competing interests
We declare that we have no financial or personal relationships which may have inappropriately influenced us in writing this article.
Authors' contributions
L.K. was the project leader; L.M. was responsible for the project design, experimental design and performed most of the experiments; R.J. performed some of the experiments; J.M.K. and M.T. were responsible for the experimental design and performed most of the wet-dry techniques and spore inoculums monitor experiments; L.M. wrote the manuscript with editorial contributions from L.K., R.J. and M.T.
References
1. Smith IM, McNamara DG, Scott PR, et al. Quarantine pests for Europe. Data sheets on quarantine pests for the European Union and for the European and Mediterranean Plant Protection Organization. 2nd ed. Wallingford, UK: CABI; 1997. [ Links ]
2. Bonants PJM, Carroll GC, DeWeerdt M, et al. Development and validation of a fast PCR-based detection method for pathogenic isolates of the citrus black spot fungus, Guignardia citricarpa. Eur J Plant Path. 2003;109:503-513. http://dx.doi.org/10.1023/A:1024219629669 [ Links ]
3. Peres NA, Harakava R, Carroll GC, et al. Comparison of molecular procedures for detection and identification of Guignardia citricarpa and G. mangiferae. Plant Dis. 2007;91:525-531. http://dx.doi.org/10.1094/PDIS-91-5-0525 [ Links ]
4. Meyer L, Sanders GM, Jacobs R, et al. A one-day sensitive method to detect and distinguish between the citrus black spot pathogen Guignardia citricarpa and the endophyte Guignardia mangiferae. Plant Dis. 2006;90:97-101. http://dx.doi.org/10.1094/PD-90-0097 [ Links ]
5. Glienke C, Pereira OL, Stringari D, et al. Endophytic and pathogenic Phyllosticta species, with reference to those associated with citrus black spot. Persoonia. 2011;26:7-56. http://dx.doi.org/10.3767/003158511X569169 [ Links ]
6. Wager VA. The black spot disease of citrus in South Africa. Sci B Dep Agric Union S Afr. 1952;303:1-52. [ Links ]
7. McOnie KC. Germination and infection of citrus by ascospores of Guignardia citricarpa in relation to control of black spot. Phytopathology. 1967;57:743-746. [ Links ]
8. Brodrick HT. Light and temperature effects on symptom development of citrus black spot disease. Citrus Series H.1.1.1(a): Farming in South Africa. Pretoria: Department of Agricultural Technical Services; 1975. [ Links ]
9. Kotzé JM. Epidemiology and control of citrus black spot in South Africa. Plant Dis. 1981;65:945-950. http://dx.doi.org/10.1094/PD-65-945 [ Links ]
10. Meyer L, Slippers B, Korsten L, et al. Two distinct Guignardia species associated with citrus in South Africa. S Afr J Sci. 2001;97:191-194. [ Links ]
11. Baayen RP, Bonants PJM, Verkley G, et al. Non-pathogenic isolates of the citrus black spot fungus. Guignardia citricarpa, identified as a cosmopolitan endophyte of woody plants, G. mangferae (Phyllosticta capitalensis). Phytopathology. 2002;92:464-477. http://dx.doi.org/10.1094/PHYTO.2002.92.5.464, PMid:18943020 [ Links ]
12. McOnie KC. The latent occurrence in citrus and other hosts of a Guignardia easily confused with G. citricarpa, the citrus black spot pathogen. Phytopathology. 1964;54:40-43. [ Links ]
13. European and Mediterranean Plant Protection Organisation. Diagnostic protocols for regulated pests: Guignardia citricarpa. EPPO Bulletin. 2003;33:271-280. http://dx.doi.org/10.1046/j.1365-2338.2003.00638.x [ Links ]
14. Wulandari NF, To-anun C, Hyde KD, et al. Phyllosticta citriasiana sp. nov., the cause of citrus tan spot of Citrus maxima in Asia. Fungal Divers. 2009;34:23-39. [ Links ]
15. Van Gent-Pelzer MPE, Van Brouwershaven IR, Kox LFF, et al. A TaqMan PCR method for routine diagnosis of the quarantine fungus Guignardia citricarpa on citrus fruit. J Phytopathology. 2007;155:357-363. http://dx.doi.org/10.1111/j.1439-0434.2007.01244.x [ Links ]
16. Glienke-Blanco C, Aguilar-Vildoso CI, Vieira MLC, et al. Genetic variability in the endophytic fungus Guignardia citricarpa isolated from citrus plants. Gen Mol Biol. 2002;25:251-255. http://dx.doi.org/10.1590/S1415-47572002000200021 [ Links ]
17. Kiely TB. Preliminary studies on Guignardia citricarpa n.sp.: The ascigerous stage of Phoma citricarpa McAlp and its relation to black spot of citrus. P Linn Soc N S W. 1948;73:249-292. [ Links ]
18. Kotzé JM. Studies on the black spot disease of citrus caused by Guignardia citricarpa Kiely, with particular reference to its epiphytology and control at Letaba. DSc thesis, Pretoria, University of Pretoria, 1963. [ Links ]
19. Truter M. Epidemiology of citrus black spot disease in South Africa and its impact on phytosanitary trade restrictions. PhD thesis, Pretoria, University of Pretoria, 2010. [ Links ]
20. Hurt RA, Qiu X, Wu L, et al. Simultaneous recovery of RNA and DNA from soils and sediments. Appl Env Microb. 2001;67:4495-4503. http://dx.doi.org/10.1128/AEM.67.10.4495-4503.2001, PMid:11571148, PMCid:93195 [ Links ]
21. Braid MD, Daniels LM, Kitts CL. Removal of PCR inhibitors from soil DNA by chemical flocculation. J Microbiol Meth. 2003;52(3):389-393. http://dx.doi.org/10.1016/S0167-7012(02)00210-5 [ Links ]
22. Tebbe CC, Vahjen W. Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and yeast. Appl Environ Microbiol. 1993;59:2657-2665. PMid:7690221, PMCid:182335 [ Links ]
23. Robe P, Nalin R, Capellano C, et al. Extraction of DNA from soil. Eur J Soil Biol. 2003;39(4):183-190. http://dx.doi.org/10.1016/S1164-5563(03)00033-5 [ Links ]
24. Qiagen DNeasy® plant handbook [homepage on the Internet]. c2006 [cited 2009 Oct 10]. Available from: http://www.qiagen.com/products/genomicdnastabilization purification/dneasyplantsystem/dneasyplantminikit.aspx [ Links ]
25. Whatman FTA® technology [homepage on the Internet]. c2007 [cited 2005 Jun 15]. Available from: http://www.whatman.com/FTANucleicAcidCollectionStorageandPurification.aspx [ Links ]
26. Epicentre Biotechnologies. SoilMasterTM DNA extraction kit [homepage on the Internet]. c2006 [cited 2007 Feb 20]. Available from: http://www.epibio.com/pdftechlit/178pl0310.pdf [ Links ]
27. Truter M, Kotzé JM, Janse van Rensburg TN, et al. A sampler to determine available Guignardia citricarpa inoculum on citrus leaf litter. Biosyst Eng. 2004;89:515-519. http://dx.doi.org/10.1016/j.biosystemseng.2004.08.018 [ Links ]
28. White TJ, Bruns T, Lee S, et al. Amplification and direct sequencing of fungal rDNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: A guide to methods and applications. San Diego, CA: Academic Press, 1990; p. 315-322. [ Links ]
29. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403-410. http://dx.doi.org/10.1006/jmbi.1990.9999, PMid:2231712 [ Links ]
30. Martin RR, James D, Levesque CA. Impacts of molecular diagnostic technologies on plant disease management. Annu Rev Phytopathol. 2002;38:207-239. http://dx.doi.org/10.1146/annurev.phyto.38.1.207, PMid:11701842 [ Links ]
31. Lievens B, Grauwet TJMA, Cammue BPA, et al. Recent developments in diagnosis of plant pathogens: A review. Rec Res Develop Microbiol. 2005;9:57-79. [ Links ]
 Correspondence to:
Correspondence to:
Linda Meyer
Postal address:Private Bag X20
University of Pretoria
Hatfield, Pretoria 0028, South Africa
Email: l.meyer@up.ac.za
Received: 24 Jan. 2011
Accepted: 19 Sept. 2011
Published: 06 Mar. 2012


{kind=link}
{kind=link}
{kind=link}