










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.108 n.1-2 Pretoria Jan. 2012
RESEARCH ARTICLE
First fungal genome sequence from Africa: a preliminary analysis
Brenda D. WingfieldI; Emma T. SteenkampII; Quentin C. SantanaI; Martin P.A. CoetzeeI; Stefan BamI; Irene BarnesI; Chrizelle W. BeukesII; Wai Yin ChanII; Lieschen de VosI; Gerda FourieII; Melanie FriendI; Thomas R. GordonIII; Darryl A. HerronII; Carson HoltIV; Ian KorfV; Marija KvasII; Simon H. MartinI; X. Osmond MlonyeniI; Kershney NaidooI; Mmatshepho M. PhashaII; Alisa PostmaI; Oleg RevaVI; Heidi RoosI; Melissa SimpsonI; Stephanie SlinskiIII; Bernard SlippersI; Rene SutherlandII; Nicolaas A. van der MerweI; Magriet A. van der NestI; Stephanus N. VenterII; Pieter M. WilkenI; Mark YandellIV; Renate ZipfelI; Mike J. WingfieldI
IDepartment of Genetics, Forestry and Agricultural Biotechnology Institute, University of Pretoria, Pretoria, South Africa
IIDepartment of Microbiology and Plant Pathology, Forestry and Agricultural Biotechnology Institute, University of Pretoria, Pretoria, South Africa
IIIDepartment of Plant Pathology, University of California, Davis, California, USA
IVEcceles Institute of Human Genetics, University of Utah, Salt Lake City, Utah, USA
VGenome Centre, University of California, Davis, California, USA
VIDepartment of Biochemistry, Bioinformatics Unit, University of Pretoria, Pretoria, South Africa
ABSTRACT
Some of the most significant breakthroughs in the biological sciences this century will emerge from the development of next generation sequencing technologies. The ease of availability of DNA sequence made possible through these new technologies has given researchers opportunities to study organisms in a manner that was not possible with Sanger sequencing. Scientists will, therefore, need to embrace genomics, as well as develop and nurture the human capacity to sequence genomes and utilise the 'tsunami' of data that emerge from genome sequencing. In response to these challenges, we sequenced the genome of Fusarium circinatum, a fungal pathogen of pine that causes pitch canker, a disease of great concern to the South African forestry industry. The sequencing work was conducted in South Africa, making F. circinatum the first eukaryotic organism for which the complete genome has been sequenced locally. Here we report on the process that was followed to sequence, assemble and perform a preliminary characterisation of the genome. Furthermore, details of the computer annotation and manual curation of this genome are presented. The F. circinatum genome was found to be nearly 44 million bases in size, which is similar to that of four other Fusarium genomes that have been sequenced elsewhere. The genome contains just over 15 000 open reading frames, which is less than that of the related species, Fusarium oxysporum, but more than that for Fusarium verticillioides. Amongst the various putative gene clusters identified in F. circinatum, those encoding the secondary metabolites fumosin and fusarin appeared to harbour evidence of gene translocation. It is anticipated that similar comparisons of other loci will provide insights into the genetic basis for pathogenicity of the pitch canker pathogen. Perhaps more importantly, this project has engaged a relatively large group of scientists including students in a significant genome project that is certain to provide a platform for growth in this important area of research in the future.
Introduction
The target genome
The Ascomycete fungus Fusarium circinatum is the causal agent of pitch canker, which is a serious disease that affects numerous Pinus species worldwide.1 The term 'pitch canker' refers to the large resinous cankers that develop on roots, trunks, branches and reproductive organs of established or mature Pinus hosts (Figure 1). On seedlings, the pathogen mainly causes root and collar rot, which are also the symptoms that were observed in South Africa when this pathogen was first detected in 1990.2,3 In contrast to the situation in other parts of the world, F. circinatum remained a nursery pathogen since this first outbreak, and it was only in 2007 that it emerged as a major pathogen in plantations planted to susceptible Pinus species.4 Apart from the losses associated with the plantation outbreaks of pitch canker, F. circinatum-related mortality during plantation establishment has been estimated to exceed R10 million annually.5 The pitch canker fungus thus represents a serious threat to the future of the pine forestry industry in this country.
Relatively little is known regarding the genetics of F. circinatum, with the bulk of knowledge at this level relating to its phylogeny and diagnostics,6,7 as well as to its population biology.8,9 Previous studies have, for example, shown that F. circinatum is a heterothallic fungus capable of both sexual and asexual reproduction. Unlike many other Ascomycete pathogens, sexual and asexual reproduction of F. circinatum have been shown in regions of the world where F. circinatum has been introduced relatively recently.10,11,12 Furthermore, studies have also shown that the fungus probably originated in Mexico or Central America and that it has been accidently introduced into pine-growing regions around the world.13 In all cases, however, these previous DNA-based studies have utilised information from either housekeeping loci or microsatelliterich regions, which in most cases represent small or limited portions of the pathogen's genome.
Whole-genome analysis procedures such as genetic linkage mapping and genome sequence comparisons have increased our understanding of the genetic basis of various biological phenomena in fungi. Well-known examples include the development of spores in Pleurotus pulmonarius14 and the development of ectomycorrhizal symbiosis in Laccaria bicolor.15 Such whole-genome approaches have also shed light on the evolution of fungal pathogenicity,16,17 which has also been particularly true for Fusarium species such as Fusarium oxysporum, Fusarium verticillioides and Fusarium graminearum18,19.The fact that the genomic data for these Fusarium species are in the public domain, and that a framework map is available for F. circinatum,20 therefore presents ideal opportunities to understand the genetic basis for pathogenicity in the pitch canker fungus.
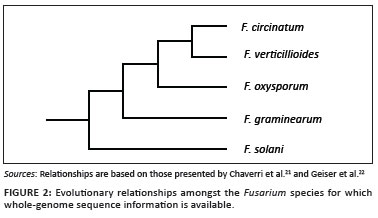
The aim of this study was to sequence, assemble and annotate the genome of F. circinatum. In addition, we present a preliminary analysis of putative gene clusters that are unique to F. circinatum and we compare three loci of this genome with the genomes of three close relatives: F. oxysporum, F. verticillioides and F. graminearum (Figure 2). From a South African perspective, this study will have significant impact - not only because the pitch canker pathogen is the first eukaryotic organism for which the entire genome has been sequenced in Africa, but also because the project strongly promotes human capacity development in the field of genome sequencing on the African continent. Furthermore, data emerging from this sequence will promote many studies concerning the pathogen and potentially lead to innovating approaches to reduce the losses that the pathogen is causing in South Africa and elsewhere in the world.
The sequence: Genome sequencing, assembly and integrity
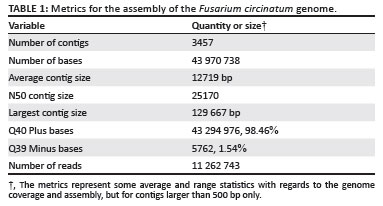
In this study we specifically targeted a F. circinatum isolate (FSP34) for which a genetic linkage map based on amplified fragment length polymorphisms is available from a previous study.20 The availability of this framework map would thus provide some higher level structure for the final genome assembly. High quality DNA was isolated23 and then sequenced on a Roche 454 GS FLX system (Life Sciences, Connecticut, USA) using the titanium chemistry by Inqaba Biotechnologies (Pretoria, South Africa). This sequencing generated a total of 500 mega bases (Mb) of DNA sequence, which comprised 1 655 231 reads (Table 1). De novo assembly of these sequences with the 454 GS assembler software package, Newbler24, resulted in 4509 contigs, the largest of which was 129 667 base pairs (bp) in length. Accordingly, the total size of the genome for F. circinatum isolate FSP34 is estimated at 43.97 Mb, which falls within the range of what has been reported for other Fusarium species. The genomes of F. graminearum, F. verticillioides and F. oxysporum are 36 Mb, 40 Mb and 60 Mb, respectively.25
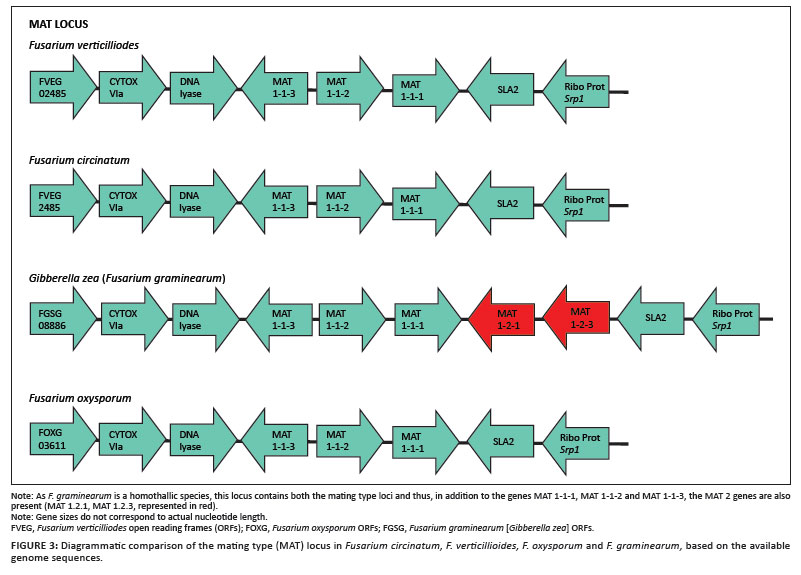
In order to confirm the integrity of the assembled F. circinatum genome, we interrogated the assembly for the presence and order of the open reading frames (ORFs) known to be encoded at the mating type (MAT) locus of this fungus. From previous research it is known that the mating type of F. circinatum isolate FSP34 is MAT-1.20,26 Within the F. circinatum assembly we thus expected to find three MAT-1 ORFs (MAT 1.1.1, MAT 1.1.2 and MAT 1.1.3) and the entire region to be flanked by genes encoding a cytoskeleton assembly control protein (SLA1) and a DNA lyase (APN1).25,27 Local Basic Local Alignment Search Tool (BLAST) analysis of the assembly indicated that a single contig (Contig00012) contained MAT-1 sequences. Examination of this 25 000 bp contig confirmed the presence of the genes, in both the same orientation and order as those found in other Fusarium species (Figure 3). This process of verification was repeated on two additional contigs (data not shown) containing genes that were of interest and also confirmed the accuracy of the assembly.
The completeness of the F. circinatum genome sequence was determined by subjecting the sequence to the CEGMA (Core Eukaryotic Genes Mapping Approach) pipeline.28 A defined set of conserved protein families known to occur in all eukaryotes was used for the analysis.28 This procedure also allows for the production of an initial set of reliable gene annotations in a eukaryotic genome, even in a draft form. The analysis revealed that the F. circinatum genome sequence assembly included the large majority (95%) of the genes common to other eukaryotes. The assembled F. circinatum genome was thus at least 95% complete. Future studies will seek to verify whether the missing genes are indeed not encoded by the pitch canker fungus.
Fusarium circinatum is a haploid fungus and the isolate sequenced was established from a single spore. Therefore, as opposed to diploid or polyploid organisms, only a single allele would be found at any particular locus in the genome. This simplifies the genome assembly process for haploid species, which generally requires less sequence coverage to produce an accurate assembly. Based on the estimated size of the F. circinatum genome and the amount of sequence information generated, an 11X sequence coverage was obtained. We were, therefore, confident that the genome of F. circinatum had been sequenced close to completeness and that the accuracy and integrity of the assembly was as good as could reasonably be expected.
Gene annotation and curation
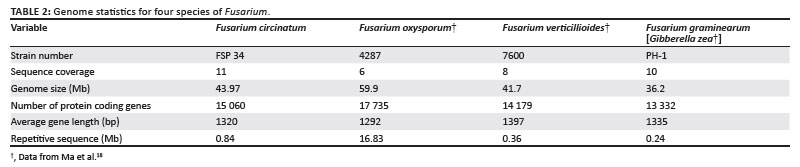
Although computer annotation of genomes has progressed substantially in the last decade, the robustness of genome annotations is still dependent on 'gene calling' programs, each of which has inherent strengths and weaknesses. Most are also designed for animals or plants with genome and gene architectures that are significantly different from those of fungi. In this study, the MAKER annotation pipeline29 was used because it is designed to particularly deal with eukaryotic genomes smaller than 100 Mb. For ab initio ORF predictions, MAKER utilised the programs Genemark ES,30 Augustus31 and SNAP.32 To streamline the ORF prediction process, the MAKER pipeline also used genome data available for F. verticillioides, F. oxysporum and F. graminearum. In addition, some expressed sequence tag (EST) sequence data were included (data not shown) to refine the accuracy of identifying the intron-exon boundaries. After several rounds of annotation to train MAKER, thereby improving its gene calling, approximately 15 000 ORFs were identified in the F. circinatum assembly (Table 2).
Whilst computer annotation programs have become substantially more sophisticated, final annotations typically need to be done manually, which currently presents the most substantial obstacle for all genome projects.33,34 In this study, we used the program Apollo35 to manually annotate and curate the F. circinatum genome. Apollo can directly utilise the sequence output from MAKER and this program also has the advantage of being relatively user friendly for biologists not familiar with computer programming.
In addition to utilising manual curation for the F. circinatum annotation, we followed the novel strategy of engaging students as annotators in the process. This approach was adopted because the skills required for curating a simple eukaryotic genome require little more than a basic degree in the biological sciences with some molecular biology focus. By following this approach we were able to achieve our second aim of promoting human capacity in the field of genome annotation in South Africa. A team of 20 graduate student volunteers was identified for this study. The students were then exposed to a 2-day training course in which the theoretical background involved in gene and genome structure was reinforced and the basic concepts and requirements of the annotation process were learned.
All the annotators were supplied with a number of contigs to curate and a support programme was implemented to assist those annotators that encountered problems. In most cases the learning curve for members of the annotation team was considerable, but tackling the annotation process in this way clearly highlighted the value of genome sequences to a biological sciences programme. The project made it possible to not only foster an appreciation of the methodologies and approaches associated with genome sequencing projects, but also provided a large number of graduate students with the opportunity to become experienced in the process of genome sequence annotation.
During the curation, each predicted ORF was compared with the predicted genes from the genomes of F. verticillioides, F. oxysporum and F. graminearum. What was immediately obvious was that about 70% of the F. circinatum ORFs were most similar to those of F. verticillioides, which is consistent with the fact that these two fungi are more closely related to one another than to the other two species (Figure 2). In many cases, when a F. circinatum ORF was not most similar to one in F. verticillioides, the dissimilarity was found to be as a result of differences in intron prediction between the two genomes. Although the ORFs in F. verticillioides have been annotated using FGENESH36 that also utilises a hidden Markov model-based algorithm to find genes, the genome of this fungus has not been subject to much manual annotation. Also, the numbers of predicted ORFs in F. circinatum and F. verticillioides differed considerably. Compared to F. circinatum, which has about 15 000 ORFs, F. verticillioides contains only about 13 500 ORFs. Of the ORFs apparently missing in F. verticillioides, a significant proportion had, in fact, not been annotated, despite the availability of EST evidence in many cases. This absence suggests that the annotation of F. verticillioides as presented on the Broad Institute website25 requires additional analyses which would probably increase the number of predicted ORFs in this genome by as much as 5%.
Inspection of the annotated output for the F. circinatum assembly revealed further discrepancies amongst the results of the different predictions programs employed by MAKER. For example, Genemark predicted 15 713 ORFs, whilst Augustus predicted 14 210 ORFs. By manually curating the annotation, it was thus possible to evaluate the various ORF prediction outputs of the pipeline in terms of intron-exon boundaries and EST evidence for F. circinatum and the other Fusarium species. After the manual curation, the F. circinatum assembly contained 15 049 predicted ORFs, with an accuracy of at least 90% for the combined gene prediction of these two programs.
From the curation it was also observed that most often the contigs terminated in intergenic regions. Although this could be ascribed to the reduced ability of the gene prediction programs to find ORFs in the absence of 3' or 5' gene signatures, the CGEMA output indicated that more than 95% of the core eukaryotic genes were present in the F. circinatum. A more likely explanation is that the assembly program Newbler was not able to assemble across DNA repeat regions, which are most often found in the intergenic regions.
Analysis of unique gene clusters
Reciprocal BLAST analyses were used to compare the predicted ORFs in the F. circinatum genome to those of the other Fusarium species. Within the resulting set of 2599 ORFs unique to F. circinatum (i.e. present in F. circinatum and absent from one or more of the other three Fusarium genomes) we identified 1031 ORFs that occurred next to each other in clusters of 4 or more. The BLAST function of the cDNA Annotation System (dCAS) v1.4.3 was then used to compare our 'unique' set of 1031 ORFs to the Pfam database (http://pfam.sanger.ac.uk/). dCAS uses the BLAST executable and BLAST databases (of which Pfam is one) to find regions of local similarity between sequences in these databases and the user's target sequence. Within the list of protein families identified amongst our 'unique' ORFs (Online Supplementary Tables 1 and 2), those with possible carbohydrate-active enzyme (CAZy) properties were identified using the CAZy database (http://www.cazy.org). The KEGG BRITE database (http://www.genome.jp/kegg/brite.html) classifications were used to group these families into classes, although a significant proportion of the ORFs could not be placed into any class using the KEGG database (Online Supplementary Table 2).
Comparison of two mycotoxin gene clusters
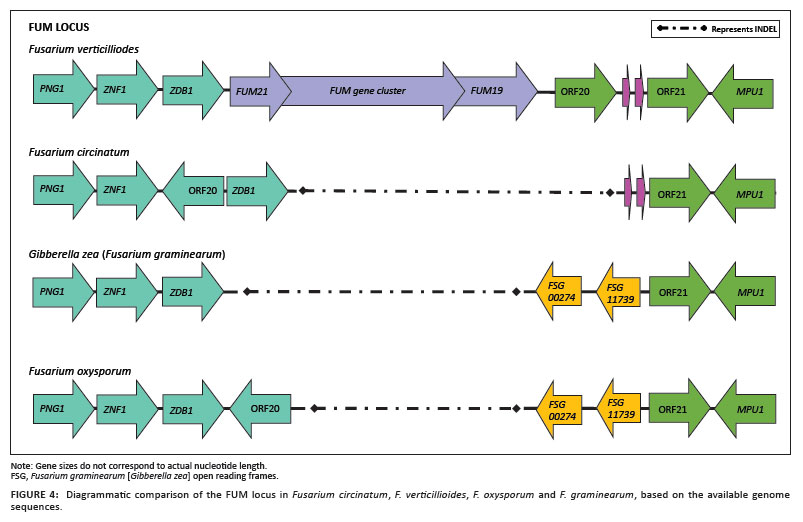
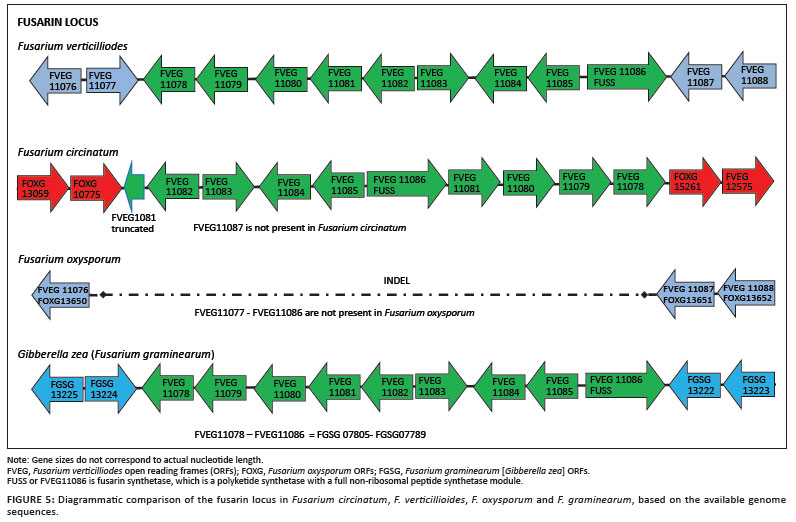
Fusarium species are widely known for the range of secondary metabolites or mycotoxins that they produce.37,38 Amongst the species for which genome sequence information is available, F. verticillioides and F. graminearum are highly toxigenic, with each capable of producing a range of mycotoxins.37,39 F. verticillioides is particularly known for producing high levels of fumonisins and fusaric acid, and F. graminearum for producing high levels of trichothecenes and zearalenone.37,39 In contrast, F. oxysporum and F. circinatum are not considered to be highly toxigenic, although some strains of F. circinatum produce beauvericin and some F. oxysporum strains produce trichothecenes and other compounds.37,38,39 The genes encoding the structural and regulatory elements involved in the biosynthesis of these toxic metabolites are usually clustered within the genomes of these species.38 In this study, we compared the genomic structure and organisation of the fumonisin and fusarin C gene clusters amongst the four Fusarium species.
The fumonisin gene cluster has been well characterised in F. verticillioides.38 By making use of this information, we were able to compare the organisation and composition of this cluster in the genomes of F. verticillioides, F. oxysporum, F. circinatum and F. graminearum (Figure 4). Our results confirm previous reports that these genes are absent in both F. circinatum and F. graminearum, neither of which has ever been shown to produce fumonisins.37,38,39,40 This gene cluster is also missing from the genome of the isolate of F. oxysporum for which the genome is available, but this locus is known to be present in another isolate (strain FRC O-1890) of the same species.41 It is interesting that one of the genes (ORF 20) flanking this locus is in a different orientation in F. oxysporum and F. circinatum and entirely missing from the F. graminearum genome. In addition, in F. circinatum ORF 20 is placed between the genes Znf1 and Zdb1, whilst these two genes are alongside each other in F. verticillioides, F. oxysporum and F. graminearum. These rearrangements and deletions thus suggest that recombination, especially in the regions flanking the cluster, determines whether fumonisin will be produced. Such recombination events could potentially facilitate horizontal transfer of this cluster amongst unrelated strains, which has been suggested to explain the patchy distribution of fumonisin production amongst lineages of Fusarium.40
A number of Fusarium species have been shown to produce the mycotoxin fusarin C, although its role in disease has not been established.37,38 Comparison of the fusarin C gene cluster in the four Fusarium species revealed its presence in F. circinatum, as well as in F. verticillioides and F. graminearum (Figure 5). Within the cluster, the gene order in F. circinatum is different from that found in F. verticillioides and F. graminearum, where the gene order is similar. In all instances the regions flanking this cluster were also unique. The fact that the locus seems to occur in a different position in the genomes of F. verticillioides, F. graminearum and F. circinatum, thus suggests that this gene cluster has been translocated more than once during the evolution of this fungal genus. In the case of F. circinatum, this translocation has been accompanied by a change in the gene order.
Discussion
This investigation had two core areas of focus. Scientifically, the aim was to sequence and annotate the genome of a fungal pathogen that is highly relevant in South Africa. The results of our preliminary comparisons represent a solid foundation of data that will significantly promote research aimed at a better understanding and management of the impact of pitch canker of pines in South Africa and elsewhere in the world. The second and equally important focus area was strongly educational, as our intention was to involve a relatively large number of students and researchers, for the first time, in a genome sequencing project. The underlying aim here was to promote an interest in this field of growing importance and to build human capacity that will contribute to similar projects in South Africa in the future.
The genome of an isolate of the pitch canker pathogen, F. circinatum, was sequenced and manually annotated. A more detailed analysis of the genome will be published during the coming year. The sequence data is available at Genbank Bioproject: PRJNA41113; ID: 41113 Locus Tag Prefix: FCIRG and on the FABI website (http://www.fabinet.up.ac.za/genomes). The F. circinatum genome, whilst being in the expected size range for a Fusarium genome, has almost 1000 more protein coding genes than its closest relative, F. verticillioides. F. oxysporum has 3000 more protein coding genes than F. verticillioides and many of these have been proposed to originate from the acquisition of lineage-specific genomic regions.19 Understanding the origin of the additional 1000 genes in F. circinatum will add to this intriguing hypothesis.
Fusarium species are well known for their production of mycotoxins. Whilst the study of mycotoxins is of particular importance for species that contaminate food and feed stocks, these toxins have also been shown to be important in plant pathogenesis.38 A comparative analysis of the F. circinatum genome with the other Fusarium genomes currently available has enabled us to determine that this fungus does not contain the fumonisin gene cluster, although the genes for the synthesis of fusarin C are present. Whether or not the pitch canker pathogen is capable of producing this compound remains to be determined. Also, the role of fusarin C has not been established in human or animal disease, nor has its role in plant pathogenesis been determined. It is perhaps most noteworthy that in the case of both these gene clusters, significant deletions or insertions and rearrangements have occurred. There is also increasing evidence of horizontal gene transfer in fungi19 and it is likely that there would be particularly strong selective pressure in terms of acquiring the ability to produce mycotoxins. Further analysis of the F. circinatum genome will focus on this possibility.
Analysis of the unique gene clusters identified in F. circinatum, whilst interesting, did not contain any significant surprises. The genome of this fungus contains a large number of proteases or peptidases, lyases and transferases that deserve further investigation. For example, these proteins could be involved in the synthesis of secondary metabolites, which in turn have the potential to be involved in pathogenesis. Amongst the four Fusarium genomes compared, F. circinatum is unique in being a gymnosperm and tree pathogen. The other three Fusarium species are pathogens typically of angiosperm monocotyledonous crops and we thus expect that F. circinatum could have a unique set of genes involved in pathogenesis.
A number of viral proteins were also found in the gene clusters, suggesting the presence of viral genomes within the F. circinatum genome. We did not filter for transposable elements and some of the genes identified as viral proteins could in fact represent elements associated with transposons. There is also precedence for the presence of both retroviruses and transposons in fungal genomes.42
The availability of the F. circinatum genome will strongly promote various projects currently being undertaken on the pathogen. For example, variation in pathogenicity was observed in the interspecies cross between F. circinatum and Fusarium subglutinans, which was used by De Vos et al.20 to produce a genetic linkage map. The availability of both the linkage map and the genome sequence will be used to study some genome regions that are potentially associated with pathogenicity. These studies will include further comparative analyses of the genome for mycotoxin biosynthetic gene clusters and other secondary metabolic gene clusters. In addition, quantitative trait loci have been identified and linked to the growth of F. circinatum (De Vos, unpublished data). With the availability of the F. circinatum genome sequence, the genetic components of these loci can now be understood and this will add to knowledge regarding the mycelial growth in fungi.
Genome sequencing is rapidly growing in importance and there is little question that this field will impact increasingly on most aspects of biology. This growth is a logical continuation of the situation that existed little more than two decades ago when the ability to sequence relatively small numbers of genes began to influence the field. The number of complete genome sequences has increased from one to close to 1000 in just 15 years.43 Apart from next generation sequencing that has substantially influenced this growth, new technological developments will ensure that this process continues, ultimately resulting in broad applications relating to genome sequences. This investigation, promoting the sequencing and annotation of a relatively small, but very significant genome, will undoubtedly come to represent a milestone in genome sequencing in South Africa.
An important element of genome sequencing is that the bulk of the work lies in annotating and interpreting the data. The entry point to this process is the laboratory component, the physical sequencing process. In South African terms, this is still relatively expensive, even for genomes as small as that of F. circinatum. However, the costs are dropping rapidly and suites of genomes, or the genomes of numerous strains of single species, are now being sequenced in consortium projects interested in comparative genomics. Whilst the wet laboratory work might be somewhat beyond the budgets of many South African laboratories, there are substantial opportunities for local scientists and students with knowledge of genome annotation to utilise genome data that is already freely available. In the future, there will be even larger volumes of genome data available for study. Some of the scientists involved in the investigation presented here will be well positioned to capitalise on this and related opportunities.
Acknowledgements
We thank the National Research Foundation (NRF) of South Africa, members of the Tree Protection Co-operative Programme, the THRIP initiative of the Department of Trade and Industry and the Department of Science and Technology (DST)/NRF Centre of Excellence in Tree Health Biotechnology and the Oppenheimer Foundation for funding.
Competing interests
We declare that we have no financial or personal relationships which may have inappropriately influenced us in writing this article.
Authors' contributions
B.D.W. was the project leader and driver behind this project; E.T.S., Q.C.S., M.J.W. and M.P.A.C. were instrumental in many of the technical aspects of the project and participated most closely in the writing of the manuscript. O.R., M.Y.,
C.H. and I.K. were the bioinformaticists on the project. The remaining authors did the bulk of the manual annotation and participated in writing the manuscript.
References
1. Wingfield MJ, Hammerbacher A, Ganley RJ, et al. Pitch canker caused by Fusarium circinatum: A growing threat to pine plantations and forests worldwide. Australas Plant Pathol. 2008;37:319-334. http://dx.doi.org/10.1071/AP08036 [ Links ]
2. Viljoen A, Wingfield MJ, Marasas WFO. First report of Fusarium subglutinans f.sp. pini on pine seedlings in South Africa. Plant Dis. 1994;78:309-312. http://dx.doi.org/10.1094/PD-78-0309 [ Links ]
3. Wingfield MJ, Coutinho TA, Roux J, Wingfield BD. The future of exotic plantation forestry in the tropics and southern hemisphere. Lessons from pitch canker. S Afr Forest J. 2002;195:79-82. [ Links ]
4. Coutinho TA, Steenkamp ET, Mongwaketsi K, Wilmot M, Wingfield MJ. First outbreak of pitch canker in a South African pine plantation. Australas Plant Pathol. 2007;36:256-261. http://dx.doi.org/10.1071/AP07017 [ Links ]
5. Mitchell RG, Wingfield MJ, Steenkamp ET, Coutinho TA. An account of Fusarium circinatum, the causal agent of the disease pitch canker on pines, with reference to Pinus patula in South Africa. South Forests. 2011;73:1-3. [ Links ]
6. Steenkamp ET, Wingfield BD, Coutinho TA, Wingfield MJ, Marasas WFO. Distinguishing Fusarium subglutinans f.sp. pini from other closely related isolates of F. subglutinans based on histone gene sequence. Appl Environ Microbiol. 1999;65:3401-3406. [ Links ]
7. Schweigkofler W, O'Donnell K, Garbelotto M. Detection and quantification of airborne conidia of Fusarium circinatum, the causal agent of pine pitch canker, from two California sites by using a real-time PCR approach combined with a simple spore trapping method. Appl Environ Microbiol. 2004;70:3512-3520. http://dx.doi.org/10.1128/AEM.70.6.3512-3520.2004 , PMid:15184151 [ Links ]
8. Britz H, Coutinho TA, Wingfield MJ, Marasas WFO, Gordon TR, Leslie JF. Fusarium subglutinans f. sp. pini represents a distinct mating population in the Gibberella fujikuroi species complex. Appl Environ Microbiol. 1999;65:1198-1201. PMid:10049883 [ Links ]
9. Santana QC, Coetzee MPA, Steenkamp ET, et al. Microsatellite discovery by deep sequencing of enriched genomic libraries. Biotechniques. 2009;46:217-223. http://dx.doi.org/10.2144/000113085, PMid:19317665 [ Links ]
10. Wikler K, Gordon TR. An initial assessment of genetic relationships among populations of Fusarium circinatum in different parts of the world. Can J Bot. 2000;78:709-717. http://dx.doi.org/10.1139/b00-044 [ Links ]
11. Britz H, Coutinho TA, Wingfield BD, Marasas WFO, Wingfield MJ. Diversity and differentiation in two populations of Gibberella circinata in South Africa. Plant Pathol. 2005;54:46-52. http://dx.doi.org/10.1111/j.1365-3059.2005.01108.x [ Links ]
12. Perez-Sierra A, Landeras E, Leon M, Berbegal M, Garcia-Jimenez J, Armengol J. Characterization of Fusarium circinatum from Pinus spp. in northern Spain. Mycol Res. 2007;111:832-839. http://dx.doi.org/10.1016/j.mycres.2007.05.009 , PMid:17662589 [ Links ]
13. Wikler K, Gordon TR, Clark SL, Wingfield MJ, Britz H. Potential for outcrossing in an apparently asexual population of Fusarium circinatum, the casual agent of pitch canker disease. Mycologia. 2000;92:1085-1090. http://dx.doi.org/10.2307/3761476 [ Links ]
14. Okuda Y, Murakami S, Matsumoto T. A genetic linkage map of Pleurotus pulmonarius based on AFLP markers, and localization of the gene region for the sporeless mutation. Genome. 2009;52:438-446. http://dx.doi.org/10.1139/G09-021 , PMid:19448724 [ Links ]
15. Martin F, Selosse MA. Prof. Dr Gopi Krishna Podila, 1957-2010 Obituary. New Phytol. 2010;186:296-297. http://dx.doi.org/10.1111/j.14698137.2010.03250.x [ Links ]
16. Soanes DM, Alam I, Cornell M, et al. Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis. PLoS ONE. 2008;3:e2300. http://dx.doi.org/10.1371/journal.pone.0002300 , PMid:18523684 [ Links ]
17. Sharpton TJ, Stajich JE, Rounsley SD, et al. Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res. 2009;19:1722-1731. http://dx.doi.org/10.1101/gr.087551.108 , PMid:19717792 [ Links ]
18. Cuomo CA, Güeldener U, Xu JR, et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007;317:1400-1402. http://dx.doi.org/10.1126/science.1143708 , PMid:17823352 [ Links ]
19. Ma LJ, Van der Does HC, Borkovich KA, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367-373. http://dx.doi.org/10.1038/nature08850, PMid:20237561 [ Links ]
20. De Vos L, Myburg AA, Wingfield MJ, Desjardins AE, Gordon TR, Wingfield BD. Complete genetic linkage maps from an interspecific cross between Fusarium circinatum and Fusarium subglutinans. Fungal Genet Biol. 2007;44:701-714. http://dx.doi.org/10.1016/j.fgb.2007.02.007 [ Links ]
21. Chaverri P, Salgado C, Hirooka Y, Rossman AY, Samuels GJ. Delimitation of Neonectria and Cylindrocarpon (Nectriaceae, Hypocreales, Ascomycota) and related genera with Cylindrocarpon-like anamorphs. Stud Mycol. 2011;68:57-78. http://dx.doi.org/10.3114/sim.2011.68.03, PMid:21523189 [ Links ]
22. Geiser DM, Lewis Ivey ML, Hakiza G, Juba JH, Miller SA. Gibberella xylarioides (anamorph: Fusarium xylarioides), a causative agent of coffee wilt disease in Africa, is a previously unrecognized member of the G. fujikuroi complex. Mycologia. 2005;97:191-201. http://dx.doi.org/10.3852/mycologia.97.1.191 , PMid:16389971 [ Links ]
23. Moller EM, Bahnwegg G, Sandermann H, Gieger HH. A simpler and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies and infected plant tissue. Nucleic Acids Res. 1992;20:6115-6116. http://dx.doi.org/10.1093/nar/20.22.6115 [ Links ]
24. 454 Sequencing home page ]homepage on the Internet[. No date ]cited 2010 Nov 28[. Available from: http://www.454.com [ Links ]
25. Broad Institute of Harvard and MIT. Fusarium Comparative Sequencing Project ]homepage on the Internet[. No date ]cited 2010 Nov 28[. Available from: http://www.broadinstitute.org [ Links ]
26. Steenkamp ET, Coutinho TA, Desjardins AE, Wingfield BD, Marasas WFO, Wingfield MJ. Gibberella fujikuroi population E associated with maize and teosinte. Mol Plant Pathol. 2001;2:215-221. http://dx.doi.org/10.1046/j.1464-6722.2001.00072.x , PMid:20573009 [ Links ]
27. Yun SH, Arie T, Kaneko I, Yoder OD, Turgeon BG. Molecular organization of mating type loci in heterothallic, homothallic, and asexual Gibberella/Fusarium species. Fungal Genet Biol. 2000;31:7-20. http://dx.doi.org/10.1006/fgbi.2000.1226, PMid:11118131 [ Links ]
28. Parra G, Bradnam K, Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061-1067. http://dx.doi.org/10.1093/bioinformatics/btm071 , PMid:17332020 [ Links ]
29. Cantarel B, Korf I, Robb SMC, et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188-196. http://dx.doi.org/10.1101/gr.6743907, PMid:18025269 [ Links ]
30. Vardges T-H, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979-1990. http://dx.doi.org/10.1101/gr.081612.108 , PMid:18757608 [ Links ]
31. Stanke M, Morgenstern B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465-W467. http://dx.doi.org/10.1093/nar/gki458 , PMid:15980513 [ Links ]
32. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004,5:59. http://dx.doi.org/10.1186/1471-2105-5-59 , PMid:15144565 [ Links ]
33. Brenner SE. Errors in genome annotation. Trends Genet. 1999,15:132-133. http://dx.doi.org/10.1016/S0168-9525(99)01706-0 [ Links ]
34. Devos D, Valencia A. Intrinsic errors in genome annotation. Trends Genet. 2001;17:429-431. http://dx.doi.org/10.1016/S0168-9525(01)02348-4 [ Links ]
35. Lewis SE, Searle SMJ, Harris N, et al. Apollo: A sequence annotation editor. Genome Biol. 2002;3:research0082-0082.14. [ Links ]
36. Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516-522. http://dx.doi.org/10.1101/gr.10.4.516, PMid:10779491 [ Links ]
37. Marasas WFO, Nelson PE, Toussoun TA. Toxigenic Fusarium species:Identity and mycotoxicology. University Park, PA: Pennsylvania State University Press; 1984. [ Links ]
38. Desjardins AE, Proctor RH. Molecular biology of Fusarium mycotoxins. Int J Food Microbiol. 2007;119:47-50. http://dx.doi.org/10.1016/j.ijfoodmicro.2007.07.024 , PMid:17707105 [ Links ]
39. Desjardins AE. Gibberella from A(venaceae) to Z(eae). Annu Rev Phytopathol. 2003;41:177-198. http://dx.doi.org/10.1146/annurev.phyto.41.011703.115501 , PMid:1265196 [ Links ]
40. Proctor RH, Plattner RD, Brown DW, Seo J-A, Lee Y-W. Discontinuous distribution of fumonisin biosynthetic genes in the Gibberella fujikuroi species complex. Mycol Res. 2004;108:815-822. http://dx.doi.org/10.1017/S0953756204000577 , PMid:15446715 [ Links ]
41. Proctor RH, Busman M, Seo J-A, Lee Y-W, Plattner RD. Fumonisin biosynthetic gene cluster in Fusarium oxysporum strain O-1890 and the genetic basis for B versus C fumonisin production. Fungal Genet Biol. 2008;45:1016-1026. http://dx.doi.org/10.1016/j.fgb.2008.02.004 , PMid:18375156 [ Links ]
42. Liu HQ, Fu YP, Jiang DH, et al. Widespread horizontal gene transfer from double-stranded RNA viruses to eukaryotic nuclear genomes. J Virol. 2010;84:11876-11887. http://dx.doi.org/10.1128/JVI.00955-10 , PMid:20810725 [ Links ]
43. Liolios K, Chen IM, Mavromatis K, et al. The genomes online database (GOLD) in 2009: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2010;38:D346-D354. http://dx.doi.org/10.1093/nar/gkp848, PMid:19914934 [ Links ]
 Correspondence to:
Correspondence to:
Brenda Wingfield
Postal address: Faculty of Natural and Agricultural Sciences, University of Pretoria
Pretoria 0002, South Africa
Email: Brenda.Wingfield@Fabi.up.ac.za
Received: 29 Nov. 2010
Accepted: 23 Nov. 2011
Published: 25 Jan. 2012
Copyright: © 2012. The Authors. Licensee: AOSIS OpenJournals. This work is licensed under the Creative Commons Attribution License.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}