










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Science
versão On-line ISSN 1996-7489
versão impressa ISSN 0038-2353
S. Afr. j. sci. vol.107 no.3-4 Pretoria Mar./Abr. 2011
http://dx.doi.org/10.4102/sajs.v107i3/4.424
RESEARCH ARTICLE
Synthesis of novel glycopolymer brushes via a combination of RAFT-mediated polymerisation and ATRP
Reda FleetI; Eric TA van den DungenI; Bert KlumpermanI,II
IDepartment of Chemistry and Polymer Science, University of Stellenbosch, South Africa
IILab of Polymer Chemistry, Eindhoven University of Technology, Eindhoven, the Netherlands
ABSTRACT
Glycopolymers (synthetic sugar-containing polymers) have become increasingly attractive to polymer chemists because of their role as biomimetic analogues and their potential for commercial applications. Glycopolymers of different structures confer high hydrophilicity and water solubility and can therefore be used for specialised applications, such as artificial materials for a number of biological, pharmaceutical and biomedical uses. The synthesis and characterisation of a series of novel glycopolymer brushes, namely poly(2-(2-bromoisobutyryloxy) ethyl methacrylate)-g-poly(methyl 6-O-methacryloyl-α-D-glucoside) (P(BIEM)-g-P(6-O-MMAGIc)), poly(2-(2-bromoisobutyryloxy) ethyl methacrylate-co-methyl methacrylate)-g-poly(methyl 6-O-methacryloyl-α-D-glucoside) P(BIEM-co-MMA)-g-P(6-O-MMAGIc), poly(2-(2-bromoisobutyryloxy) ethyl methacrylate-b-methyl methacrylate)-g-poly(methyl 6-O-methacryloyl-α-D-glucoside) P(BIEM-b-MMA)-g-P(6-O-MMAGIc) and poly(4-vinylbenzyl chloride-alt-maleic anhydride)-g-poly(methyl 6-O-methacryloyl-α-D-glucoside) (P(Sd-alt-MAnh)-g-P(6-O-MMAGIc)) are described in this paper. Reversible addition-fragmentation chain transfer (RAFT)-mediated polymerisation was used to synthesise four well-defined atom transfer radical polymerisation (ATRP) macroinitiators (the backbone of the glycopolymer brushes). These ATRP macroinitiators were subsequently used in the 'grafting from' approach (in which side chains are grown from the backbone) to prepare high molar mass and low polydispersity index glycopolymer brushes with different grafting densities along the backbone. The number average molar mass of the glycopolymer brushes was determined using size exclusion chromatography with a multi-angle laser light scattering detector and further structural characterisation was conducted using 1H-nuclear magnetic resonance spectroscopy. The results confirmed that glycopolymer brushes were successfully synthesised via a combination of RAFT-mediated polymerisation and ATRP.
Introduction
Despite a solid history in chemistry in general, polymer science has been largely overlooked on the African continent. It was already in the 1960s that Ronald D. Sanderson recognised this omission. After a PhD study at the University of Akron (USA), he returned to South Africa in 1970. With very little support from established academics, he started to pursue his dream to create a Polymer Institute of international standing. From 1978 he steadily built up the Institute with increasing numbers of students, and by attracting a number of international collaborators. Sanderson made large contributions to membrane research for water purification, a field in which he collaborated with Ed Jacobs. In collaboration with his former student Albert van Reenen, Sanderson contributed to the field of polyolefins, which is of obvious importance to the largest chemical company in South Africa, Sasol. Characterisation of polyolefins was recently boosted in Stellenbosch as a result of the arrival of Harald Pasch as the Sasol Chair of Polymer Characterisation. In addition to the polyolefin research, there is currently a strong effort in nanostructured polymer materials at Stellenbosch University. Peter Mallon has a strong focus on this field, where polymer synthesis and advanced polymer processing methods, such as electrospinning, are employed next to advanced characterisation techniques. Bert Klumperman holds a South African Research Chair on Advanced Macromolecular Architectures. His research is centered around polymer synthesis, where the focus is increasingly on polymers for biomedical applications. The present contribution shows an example from work on a specific class of advanced macromolecular architectures, i.e. polymer brushes. Polymer brushes have generated much interest because of their unique properties and their ability to alter the surface properties of materials.1 It is known that the solution and bulk properties of polymer brushes are significantly influenced by their chain architecture.2 Their properties depend on a variety of molecular parameters, including the degree of polymerisation of the backbone and side chains, the grafting density, the main chain topology and the chemical composition.3
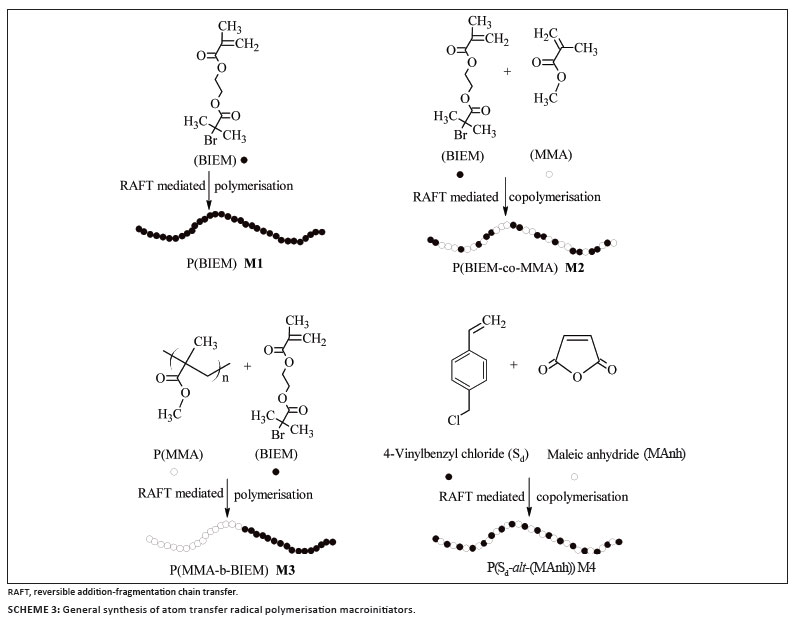
A polymer brush is defined as an assembly of polymer chains which are densely tethered by one end to a surface or an interface.4 As a result of high steric crowding, the chains are forced to stretch away from the surface to avoid segmental overlap.1,4,5 The architecture of polymer brushes can be varied by being densely or loosely grafted, having flexible or stiff side chains, or being homopolymers or copolymers.5,6 Three synthetic routes for the preparation of polymer brushes are described in the literature3,6,7,8: (1) 'grafting onto' (attachment of the side chains to the backbone), (2) 'grafting through' (homopolymerisation and copolymerisation of macromonomers) and (3) 'grafting from' (growing side chains from the backbone). To achieve high grafting density, the 'grafting from' approach via atom transfer radical polymerisation (ATRP) has proven particularly beneficial.9,10,11,12
The ATRP macroinitiator can be prepared in different ways. For example, Hawker et al.13 and Beers et al.14 prepared ATRP macroinitiators by nitroxide-mediated polymerisation (NMP) and ATRP, respectively. Another interesting example is the work reported by Venkatesh et al.3, who used the reversible addition-fragmentation chain transfer (RAFT) technique to prepare ATRP macroinitiators without the need to resort to protecting group chemistry on the ATRP initiator moieties.
Recently, glycopolymers (synthetic sugar-containing polymers) have become increasingly attractive to polymer chemists because of their role as biomimetic analogues and their potential for commercial applications.15,16,17 Glycopolymers of different structures confer high hydrophilicity and water solubility and can therefore be used for specialised applications, such as artificial materials for a number of biological, pharmaceutical and biomedical uses.18,19,20,21 Glycopolymers can be prepared via two different approaches. The first method involves the polymerisation of sugar-based monomers, whereas the second method entails the chemical modification of preformed polymers with sugar-containing reagents.22,23 Glycopolymers can be defined in a general sense as synthetic polymers possessing a non-carbohydrate backbone that carry carbohydrate (sugar) moieties as pendant or terminal groups.15,24
In this work the glycomonomer was synthesised first, using an enzymatic approach to regioselectively functionalise the primary hydroxyl group of methyl α-D-glucoside. The glycomonomer was subsequently polymerised. The introduction of a vinyl functionality in this position (primary hydroxyl group) has been found to eliminate the biological activity of the resulting glycopolymer.25 Various ATRP macroinitiators were synthesised via RAFT-mediated polymerisation, without the need to use protecting group chemistry for the ATRP-initiating moieties. Glycopolymer brushes with poly(methyl 6-O-methacryloyl-α-D-glucoside) (P(6-O-MMAGlc)) side chains were then prepared via ATRP using the 'grafting from' approach. The polymer brushes were characterised via 1H-nuclear magnetic resonance (NMR) spectroscopy and size exclusion chromatography (SEC).
Methods and materials
Chemicals
2-Hydroxyethyl methacrylate 98% (Sigma-Aldrich, Johannesburg, South Africa), triethylamine 99.5% (Sigma-Aldrich), dichloromethane 98% (Merck, Johannesburg, South Africa), 2-bromoisobutyryl bromide 98% (Sigma-Aldrich), sodium hydrogen carbonate 99% (Fluka, Johannesburg, South Africa), sodium chloride 98% (Sigma-Aldrich), distilled deionised water was obtained from a Millipore Milli-Q purification system (Cape Town, South Africa), magnesium sulphate (anhydrous) 99% (Saarchem, Johannesburg, South Africa), n-propylamine 99% (Alfa Aesar, Johannesburg, South Africa), pyridine-2-carboxaldehyde 99% (Sigma-Aldrich), diethyl ether 99.7% (Sigma-Aldrich), methyl α-D-glucoside 99% (Sigma-Aldrich), Novozym® 435 (an immobilized lipase; Novozymes (Pty) Ltd, Johannesburg, South Africa), vinyl methacrylate 98% (Sigma-Aldrich), anhydrous acetonitrile 99.8% (Sigma-Aldrich), methanol 98% (Alfa Aesar), ethyl acetate (Sasol Class 3), ethanol (Kimix CP, Cape Town, South Africa), hexane (Kimix CP), p-xylene 99% (Merck), 1,4-dioxane 99% (Saarchem uniLAB), methyl ethyl ketone 99.7% (Sigma-Aldrich), maleic anhydride 99% (Acros Organics, Cape Town, South Africa), 4-vinylbenzyl chloride 90% (Sigma-Aldrich), copper(I)bromide 99% (Sigma-Aldrich), ethyl a-bromoisobutyrate 98% (Fluka) and N,N-dimethylformamide 97% (Fluka) were all used as received without further purification. Methyl methacrylate (MMA) 99% (Sigma-Aldrich) was purified by passing through a column of basic aluminium oxide to remove the inhibitor. The initiator 2,2'-azobis(isobutyronitrile) (AIBN, Riedel de Haën, Johannesburg, South Africa) was re-crystallised twice from ethanol and dried under vacuum before use.
Characterisation
1H-NMR and 13C-NMR spectra were obtained from a Varian VXR 400 MHz instrument, or from a Varian Unity Inova 600 MHz spectrometer (Agilent, Palo Alto, California, USA). Depending on the solubility of the synthesised compounds, deuterated chloroform (CDCl3) or deuterated dimethyl sulphoxide (DMSO-d6) was used as the solvent. All chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane (TMS), used as an internal standard (δ = 0 ppm).
Molar masses and molar mass distributions were measured using SEC. The SEC instrument consisted of a Waters 117 plus auto-sampler, a Waters 600E system controller (run by Waters Millennium32 software26) and a Waters 610 fluid unit (Waters Corporation, Milford, MA, USA). A Waters 410 differential refractometer and a Waters 2487 dual wavelength absorbance detector were used. A laser photometer miniDAWN (Wyatt Technology Corporation, Santa Barbara, CA, USA) multi-angle laser light scattering (MALLS) detector with ASTRA software27 was used. The system was equipped with a 50 mm x 8 mm guard column and three 300 mm x 8 mm linear columns (Gram (PSS), 3 x 103 Å, 3 x 102 Å and 3 x 103 Å pore size; 10-μm particle size). N,N-dimethylacetamide (HPLC grade, 0.03% w/v, LiCl, 0.05% butylated hydroxytoluene) was used as eluent at a flow rate of 1 mL/min, while the column oven was kept at 40 ºC and 100 µL of 5 mg/mL polymer solution was injected. The system was calibrated with narrow PMMA standards ranging from 800 g/mol to 2 x 106 g/mol. All molar masses are reported as PMMA equivalents.
Atomic force microscopy (AFM) measurements were performed with an Easy Scan II AFM (Nanosurf AG, Liestal, Switzerland). The microscope was operated in tapping mode at a resonance frequency of 360 kHz at ambient conditions.
Synthesis of 2-cyanoprop-2-yl dithiobenzoate
The synthesis of 2-cyanoprop-2-yl dithiobenzoate (CIPDB) was carried out according to the method of Moad et al. 28 and purified by successive column chromatography with silica as the stationary phase and hexane:diethyl ether (9:1) as the mobile phase. After removal of solvent under reduced pressure, the product was stored below -10 ºC. The purity of the RAFT agent was estimated via 1H-NMR spectroscopy to be 96%.
Synthesis of 2-(2-bromoisobutyryloxy) ethyl methacrylate
The synthesis of 2-(2-bromoisobutyryloxy) ethyl methacrylate (BIEM) was carried out according to the procedure described previously (Scheme 1).3,29 Briefly, a solution of 2-hydroxyethyl methacrylate (HEMA, 10.0 g, 0.07 mol) and triethylamine (TEA, 15.6 g, 0.15 mol) in dichloromethane (DCM, 50 mL) was stirred at 0 ºC for 45 min under argon. A solution of 2-bromoisobutyryl bromide (21.2 g, 0.09 mol) in DCM (25 mL) was added dropwise over a period of 30 min. The mixture was stirred at 0 ºC for 6 h under an argon atmosphere and then filtered to remove the precipitated solids. The solids were washed with DCM and the filtrate was then washed twice with deionised water (100 mL), a 0.5 M NaHCO3 solution (100 mL) and a saturated NaCl solution (100 mL). Sodium sulphate was added to remove traces of water, and then filtered off. The DCM was removed at 25 ºC under vacuum. The purity of the obtained product was checked by 1H-NMR spectroscopy and estimated to be 99%. The yield obtained was 94%.
1H-NMR (CDCl3) δ (ppm) from TMS: 6.10 (m, 1H), 5.56 (m, 1H, CH2=C), 4.39 (m, 4H, -O-CH2-CH2-O-), 1.91 (q, 3H, α-CH3), 1.89 (s, 6H, -C(Br)(CH3)2). 13C-NMR (CDCl3) δ (ppm) from TMS: 171.5 (-O-C=O), 167 (O=C-O-), 135.69 (CH2=C), 126.1 (CH2=C), 63.44 (CH2-O), 61.93 (O-CH2), 55.43 (-C(Br)(CH3)2), 30.5 (-C(Br)(CH3)2), 18.67 (α-CH3). ESI-MS: m/z calculated for C10H16BrO4: 280.12 g/mol, found: 281 g/mol (M+H+).
Preparation of N-(n-propyl)-2-pyridylmethanimine
The synthesis of N-(n-propyl)-2-pyridylmethanimine (n-Pr-1) was carried out according to the method of Haddleton et al.30 An excess of n-propylamine (2.95 g, 0.05 mol) was added dropwise to a cooled solution of pyridine-2-carboxaldehyde (4.5 g, 0.042 mol) in diethyl ether (5 mL) while being stirred. After complete addition of the amine, anhydrous magnesium sulphate (4 g) was added and the slurry stirred for 5 h at 25 ºC. The solution was filtered, and the solvent removed to afford a gold/yellow oil. The yield obtained was 5.9 g (94.7%). 1H-NMR (CDCl3) δ (ppm) from TMS: 8.60 (m, 1H), 8.38 (s, 1H), 7.97 (m, 1H), 7.69 (m, 1H), 7.27 (m, 1H), 3.62 (t, 2H), 1.71 (m, 2H), 0.94 (t, 3H).
Synthesis of methyl 6-O-methacryloyl-α-D-glucoside
Methyl 6-O-methacryloyl-α-D-glucoside (6-O-MMAGlc, monomer) was prepared according to the method of Albertin et al.15,31 with some modifications (Scheme 2). A conical flask was charged with methyl α-D-glucoside (8.0 g, 0.041 mol), Novozym® 435 (4.0 g), vinyl methacrylate (4.4 g, 0.039 mol) and dry acetonitrile (40 mL). Novozym® 435 was chosen as the catalyst because of its ability to catalyse regioselective esterification and transesterification reactions in a number of organic solvents.
The flask was sealed with a stopper and the suspension stirred at 200 rpm and 50 ºC for 7 days before the reaction was stopped by filtering off the enzyme. The filtrate was washed with methanol (100 mL) and the collected organic phases were rotary evaporated to dryness to yield a yellow-brown syrup. Ethyl acetate (100 mL) was then added to the syrup in order to precipitate the unreacted methyl α-D-glucoside. The product was purified by column chromatography on silica as the stationary phase using volume ratios of 7:2:1 of ethyl acetate:hexane:ethanol as the mobile phase. The solvent was removed from the collected fractions via a rotary evaporator under reduced pressure at room temperature (to avoid polymerisation). A transparent syrup of 6-O-MMAGlc monomer was obtained in 64% yield with respect to methyl α-D-glucoside.
1H-NMR (CDCl3) δ (ppm) from TMS: 1.92 (s, 3H, H-11), 3.33 (d, 1H, H-4), 3.37 (s, 4H, H-7 and dd, H-2), 3.52 (dd, 1H, H-3), 3.72 (m, 1H, H-5), 4.09 (dd, 1H, H-6), 4.31 (dd, 1H, H-6), 4.43 (d, 1H, H-1), 4.72 (d, 1H, H-10), 5.55 (t, 1H, H-10). 13C-NMR (CDCl3) δ (ppm) from TMS: 18.28 (C-11), 55.08 (C-7), 64.04 (C-6), 69.73 (C-5), 70.45 (C-4), 71.97 (C-2), 74.11 (C-3), 99.40 (C-1), 126.12 (C-10), 136.02 (C-9), 167.57 (C-8). ESI-MS m/z calculated for C11H22O7N, 280.14 g/mol, found 280 g/mol (M + NH4+).
Polymerisation procedures
All polymerisations were carried out in a pear-shaped 50-mL Schlenk flask heated in an oil bath. The polymerisation solution was degassed using a minimum of three freeze-pump-thaw cycles followed by the introduction of high purity argon. The target molecular weight was calculated from the following equation

where [M]0 and [RAFT]0 are the initial concentrations of the monomer and the RAFT agent, MWM and MWRAFT are the molar masses of the monomer and the RAFT agent, x is the fractional conversion and Mn,th is the theoretical number average molar mass of the formed polymer.
RAFT-mediated polymerisation of BIEM
A stock solution was prepared by accurately weighing the monomer BIEM (2 g, 7.0 x 10-3 mol), RAFT agent CIPDB (0.02 g, 9.03 x 10-5 mol), AIBN (2.9 x 10-3 g, 1.8 x 10-5 mol) and the solvent p-xylene (4 g) in a Schlenk flask. The stock solution was degassed via three freeze-pump-thaw cycles, immersed in a thermostatted oil bath preheated to 60 ºC and stirred using a magnetic stirrer. After 24 h the P(BIEM) was isolated by precipitation in methanol. After filtration the polymer was dried under vacuum for 24 h. The polymer was analysed by SEC and 1H-NMR spectroscopy and the molar mass was found to be Mn = 13.0 x103 g/mol and the PDI was 1.12.
RAFT-mediated copolymerisation of BIEM and MMA
A stock solution of BIEM (1 g, 3.5 x 10-3 mol), MMA (0.35 g, 3.5 x 10-3 mol), RAFT agent CIPDB (0.014 g, 6.2 x 10-5 mol), AIBN (0.002 g, 1.26 x 10-5 mol), and 1,4-dioxane (3 g) was prepared in a Schlenk flask and degassed by three freeze-pump-thaw cycles. The Schlenk flask was immersed in a thermostatted oil bath preheated to 60 ºC. After 24 h of reaction under magnetic stirring, P(BIEM-co-MMA) was isolated by precipitation in methanol. After filtration the polymer was dried under vacuum for 24 h. The polymer was analysed using SEC and 1H-NMR spectroscopy; the molar mass was determined to be Mn = 10 x 103 g/mol and the PDI = 1.14. The copolymer resulted in a random distribution in composition along the backbone as a result of the reactivity ratios.6
RAFT-mediated polymerisation of BIEM using a P(MMA) macroRAFT agent
A 50-mL Schlenk flask was charged with BIEM (1 g, 3.5 x 10-3 mol), AIBN (0.003 g, 1.8 x 10-5 mol) and 1,4-dioxane (5 g). A stock solution of P(MMA) macroRAFT agent (0.35 g, Mn = 5.0 x 103 g/mol, PDI = 1.12, prepared according to a procedure described previously26) in 1,4-dioxane was added to the mixture in the Schlenk flask. The solution was then degassed via three freeze-pump-thaw cycles, after which the flask was immersed in a thermostatted oil bath preheated to 60 ºC. After 24 h reaction under magnetic stirring the P(MMA-b-BIEM) was isolated by precipitation in methanol. The product was dried in a vacuum oven for 24 h and analysed by SEC and 1H-NMR spectroscopy: the molar mass was Mn = 9.0 x 103 g/mol and the PDI = 1.21.
RAFT-mediated copolymerisation of 4-vinylbenzyl chloride and maleic anhydride
In a typical RAFT-mediated polymerisation, a magnetic stirrer was placed in a 50-mL Schlenk flask along with maleic anhydride (MAnh) (0.98 g, 0.01 mol), 4-vinylbenzyl chloride (Sd) (2 g, 0.01 mol), RAFT agent CIPDB (0.03 g, 1.36 x 10-4 mol), AIBN (0.004 g, 2.7 x 10-5 mol) and methyl ethyl ketone (MEK) (6 g). The solution was degassed by three freeze-pump-thaw cycles. The Schlenk flask was immersed in a thermostatted oil bath preheated to 60 ºC. After 24 h of reaction under magnetic stirring, P(Sd-alt-MAnh) was isolated by precipitation in diethyl ether. The copolymer was dried in a vacuum oven for 24 h and analysed by SEC and 1H-NMR spectroscopy: the molar mass was Mn = 15.0 x 103 g/mol and the PDI = 1.16. The synthesis of P(Sd-alt-MAnh) resulted in the formation of an alternating copolymer.32
Synthesis of glycopolymer brushes
A series of glycopolymer brushes with similar molecular weight of the side chains and different grafting densities were prepared by using the 'grafting from' approach. All polymerisations were carried out in a pear-shaped 50-mL Schlenk flask heated in an oil bath. The polymerisation mixture was degassed by a minimum of three freeze-pump-thaw cycles followed by the introduction of high purity argon. Monomer conversion for the graft polymerisations was determined gravimetrically. The degree of polymerisation (DP) of the side chains was calculated using the following equation, based on monomer conversion and assuming quantitative initiation from each Br or Cl atom:

where [M]0 is the initial monomer concentration and [I]0 is the initial initiator concentration.
Synthesis of P(BIEM)-g-P(6-O-MMAGIc
A stock solution for the graft polymerisation was prepared in a 50-mL Schlenk flask by accurately weighing P(BIEM) (0.04 g, 0.14 x 10-3 mol of initiating α-bromoester group, obtained from 1H-NMR spectroscopy), Cu(I)Br (0.01 g, 0.07 x 10-3 mol) and n-Pr-1 ligand (0.02 g, 0.14 x 10-3 mol). The mixture was stirred in N,N-dimethylformamide (DMF, 4 g) for 15 min to dissolve the macroinitiator completely. A solution of 6-O-MMAGlc monomer (2 g, 7.0 x 10-3 mol) in DMF (2 g) was then added and the reaction mixture was degassed by three freeze-pump-thaw cycles followed by the introduction of high purity argon. The flask was sealed with a rubber septum and placed in a preheated thermostatted oil bath at 60 ºC. The polymerisation was stopped after 1 h by cooling to room temperature and opening the flask to air. The polymerisation solution, which was very viscous even at low conversions, was diluted with DMF and passed three times through a column of neutral aluminium oxide to remove any trace of the catalyst. This was followed by precipitation in diethyl ether twice to remove unreacted monomer. The yield was 0.8 g of isolated polymer. The product was characterised by 1H-NMR spectroscopy and SEC. According to SEC using poly(methyl methacrylate) (PMMA) calibration, the molar mass was Mn = 85.0 x 103 g/mol and the PDI = 1.21. The monomer conversion was 40%.
Synthesis of P(MMA-co-BIEM)-g-P(6-O-MMAGIc)
P(MMA-co-BIEM) (0.09 g, 0.14 x 10-3 mol of initiating α-bromoester group, obtained from 1H-NMR spectroscopy), Cu(I)Br (0.01 g, 0.07 x 10-3 mol), n-Pr-1 ligand (0.02 g, 0.14 x 10-3 mol) and DMF (4 g) were added to a 50-mL Schlenk flask. A solution of 6-O-MMAGlc monomer (2 g, 7.0 x 10-3 mol) in DMF (2 g) was then added to the reaction mixture. The flask was sealed with a rubber septum and placed in a preheated thermostatted oil bath at 60 ºC. The polymerisation was stopped after 90 min by cooling to room temperature and opening the flask to air. The polymerisation solution was diluted with DMF and passed three times through a column of neutral aluminium oxide to remove any trace of the catalyst. This was followed by precipitation in diethyl ether twice to remove unreacted monomer. The yield was 1.1 g of isolated polymer; and the monomer conversion was 55%. According to SEC using PMMA calibration, the molar mass was Mn = 55.0 x 103 g/mol and the PDI = 1.25.
Synthesis of P(MMA-b-BIEM)-g-P(6-O-MMAGIc)
A magnetic stirrer was placed in a 50-mL Schlenk flask together with P(MMA-b-BIEM) (0.08 g, 0.14 x 10-3 mol of initiating α-bromoester group, obtained from 1H-NMR spectroscopy), Cu(I)Br (0.01 g, 0.07 x 10-3 mol), n-Pr-1 ligand (0.02 g, 0.14 x 10-3 mol) and DMF (4 g). The solution was stirred until a homogenous mixture was obtained. A solution of 6-O-MMAGlc monomer (2 g, 7.0 x 10-3 mol) in DMF (2 g) was then added to the reaction mixture. The reaction mixture was degassed by three freeze-pump-thaw cycles followed by the introduction of high purity argon. The flask was sealed with a rubber septum and placed in a preheated thermostatted oil bath at 60 ºC. The polymerisation was stopped after 90 min by cooling to room temperature and opening the flask to air. After diluting the viscous polymer solution with DMF, the solution was passed three times through a neutral aluminium oxide column to remove the catalyst. This was followed by precipitation in diethyl ether twice to remove unreacted monomer. The yield was 0.9 g of isolated polymer and the 6-O-MMAGlc conversion was 45%. SEC with PMMA calibration was used to determine the molar mass (Mn = 47.0 x 103 g/mol) and the PDI (1.29).
Synthesis of P(Sd-alt-MAnh)-g-P(6-O-MMAGIc)
A 50-mL Schlenk flask was charged with P(Sd-alt-MAnh) (0.05 g, 0.15 x 10-3 mol of initiating chloride atoms, obtained from 1H-NMR spectroscopy), Cu(I)Br (0.01 g, 0.07 x 10-3 mol), n-Pr-1 ligand (0.02 g, 0.15 x 10-3 mol) and DMF (4 g). A solution of 6-O-MMAGlc (2 g, 7.0 x 10-3 mol) in DMF (2 g) was then added to the reaction mixture. The polymerisation was stopped after 90 min by cooling to room temperature and opening the flask to air. This was followed by precipitation in diethyl ether twice to remove unreacted monomer. The yield was 0.7 g of isolated polymer and the monomer conversion was 35%. SEC with PMMA calibration was used to determine the molar mass (63.0 x 103 g/mol) and PDI (1.32).
Results and discussion
Synthesis and characterisation of ATRP macroinitiators and their corresponding glycopolymer brushes
The RAFT-mediated homopolymerisation of BIEM, the RAFT-mediated copolymerisation of BIEM and MMA and the RAFT-mediated copolymerisation of Sd and MAnh were investigated for the preparation of ATRP macroinitiators. The synthesis of a macroinitiator with a low PDI is crucial because the molar mass distribution (MMD) of the polymer brushes is largely dependent on the MMD of the backbone.2,3 CIPDB was used as the RAFT agent for the synthesis of the macroinitiators because it is known from the literature that it controls the polymerisation of MMA because of its high chain transfer coefficient coupled with the fact that the 2-cyanoprop-2-yl leaving group is a good initiating species for MMA polymerisation.28 It was expected that, by using RAFT-mediated polymerisation for the synthesis of ATRP macroinitiators, the need to use protecting group chemistry for the ATRP initiator could be avoided.3
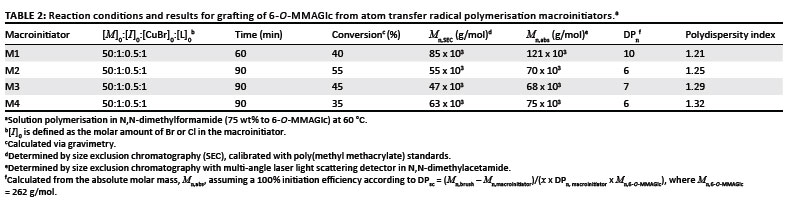
ATRP macroinitiators with a different distribution of initiating species along the backbone were prepared via RAFT-mediated polymerisation according to Scheme 3. Because these macroinitiators were going to function as the backbone for the polymer brushes, they were extensively characterised in order to provide information on the initiator density (Table 1). The actual number of initiating groups along the backbone was calculated from the DP of the backbone and the molar fraction of BIEM units along the backbone, which was determined by 1H-NMR spectroscopy. The 1H-NMR spectrum of macroinitiator M1 in Figure 1a clearly shows two typical peaks at 4.22 ppm and 4.38 ppm (peak b), which represent the methylene protons between the two ester groups of the macroinitiator.2 For the macroinitiators M2 and M3 (Figures 1b and 1c, respectively), the integration area of the -OCH3 protons at 3.6 ppm (peak c) from MMA and the four protons from BIEM at 4.22 ppm and 4.38 ppm (peak b) were compared to determine the final copolymer composition.6 The ratio between PBIEM and PMMA for the macroinitiators M2 and M3 was calculated to be 45:55 and 51:49, respectively.
The 1H-NMR spectrum of macroinitiator M4 is shown in Figure 1d. The poorly resolved resonance peak at 6.1 ppm - 7.5 ppm is ascribed to the four protons of the benzene ring of 4-vinylbenzyl chloride, while the peak at 2.31 ppm is associated with the protons of maleic anhydride.33,34 The integral ratio of the respective areas indicated that the composition of 4-vinylbenzyl chloride to maleic anhydride was approximately 49:51.
When polymer brushes are synthesised by the 'grafting from' approach via radical polymerisation, it is necessary to ensure a low concentration of active species. This is because of the tendency for termination to take place via both intramolecular and intermolecular coupling, which leads to cross-linked polymers or polymers with multimodal distributions.11 Therefore, low temperatures and low active species (Cu(I)) concentration were used to reduce the concentration of radicals during the polymerisation. An increase in the viscosity at higher conversions was avoided by using a solvent to monomer weight ratio of 3:1. Glycopolymer brushes were synthesised with an approximately similar DP of side chains by grafting 6-O-MMAGlc from the four macroinitiators via ATRP.
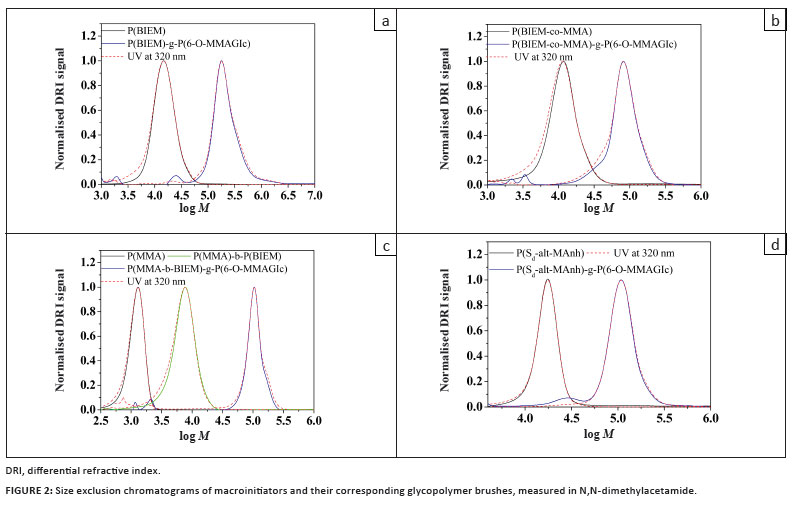
The experimental molar masses and MMDs of the macroinitiators and their corresponding glycopolymer brushes were investigated by SEC. Figure 2 shows that each chromatogram exhibits a narrow monomodal MMD (PDI < 1.4). The SEC traces shift to higher molar mass, indicating that high molar mass glycopolymer brushes were formed. There was no significant tailing or shoulder formation observed, indicating that intermolecular coupling reactions (brush-brush coupling) during the polymerisation were negligible.35 The synthesis of the glycopolymer brushes proceeded in a controlled fashion as the PDI of all the glycopolymer brushes remained comparable to that of the macroinitiators.6 It was noticed that the polymerisation of 6-O-MMAGlc was very fast and it went to relatively high conversion, as can be seen in Table 2. This response was expected, because 6-O-MMAGlc acts as a reducing agent, reducing the deactivator to regenerate the activator, and therefore the rate of polymerisation is enhanced.36
The Mn values obtained from SEC using refractive index detection relative to PMMA standards are apparent values. The lower values of the experimental Mn for the glycopolymer brushes could be because the glycopolymer brushes have a lower hydrodynamic volume than the equivalent linear polymers. As a result, the glycopolymer brushes elute later during SEC compared to their linear counterparts. In order to obtain a more accurate estimate of Mn and PDI of the glycopolymer brushes, SEC with a MALLS detector was performed in N,N-dimethylacetamide. The dn/dc value used (0.092 mL/g) was based on the composition of the side chains (P(6-O-MMAGlc)) as they comprised the bulk of the material (> 95%).14 The absolute molar masses obtained from these measurements are shown in Table 2. It is clear that the true molar masses are significantly higher than the apparent ones. This finding is in accordance with the general phenomenon that polymer brushes are more compact than linear chains with identical molar mass.3
The presence of the RAFT agent end group can be determined using dual detection for SEC. Ultraviolet (UV) and refractive index (RI) detectors were used to determine whether the polymer chains contained the RAFT agent as an end group. The UV detector was set at 320 nm, as the thiocarbonyl thio moiety (-S(C=S)-) of the RAFT agent strongly absorbs at this wavelength.37 Overlay of the two signals indicates whether the RAFT agent functionality is homogeneously or heterogeneously distributed throughout the molar mass distribution. The graphs in Figure 2 show that the RAFT agent moiety was homogenously distributed throughout the molecular weight distribution. The delay between the RI and the UV detectors was compensated for so that both signals overlapped at their peak maximum. At low molar mass the observed UV signal was very strong because the chains were small, resulting in a high concentration of RAFT agent per mass of chain.38
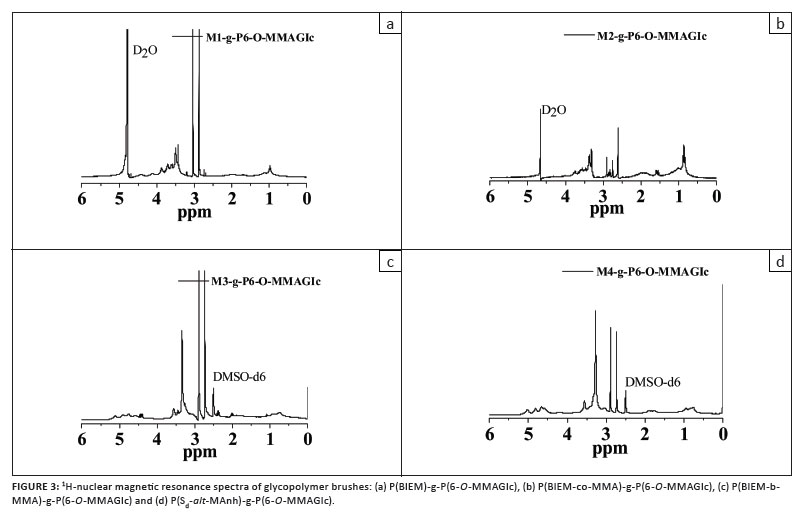
Figure 3 shows the 1H-NMR spectra of the glycopolymer brushes. A broad peak attributed to anomeric hydroxyl groups of the sugar moieties (3.2 ppm - 3.8 ppm in D2O and 2.9 ppm - 3.6 ppm in DMSO-d6) appears, and the characteristic peaks for the macroinitiators remain visible in the spectra. This demonstrates the successful formation of molecular brushes with glycopolymer side chains.
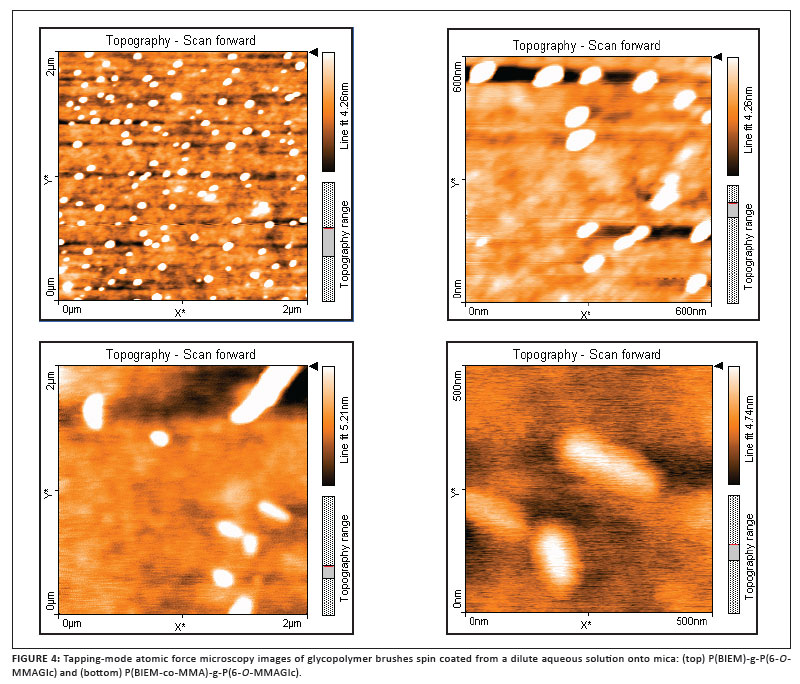
Visualisation of glycopolymer brushes by AFM
The glycopolymer brushes were further characterised by AFM in order to visualise the polymer morphology. Previous studies showed that molecular brushes could be visualised as single molecules using tapping mode AFM.7,39 Even the backbone and the side chains were clearly observed as a result of the high spatial resolution and strong material contrast of tapping mode AFM.7,40 All samples for AFM studies were prepared by spin casting from a dilute aqueous solution with the concentration varying from 0.1 mg/mL to 0.2 mg/mL. Polymers were spin-coated at room temperature at 2000 rpm on freshly cleaved mica. Figure 4 shows the AFM images of the P(BIEM)-g-P(6-O-MMAGIc) and P(BIEM-co-MMA)-g-P(6-O-MMAGIc) glycopolymer brushes.
In Figure 4, only islands of polymer molecules are visible; single molecules are not observed. This finding may be attributed to the fact that the brushes are not completely stretched because of the low DP of the backbone. We therefore speculate that the backbone tends to coil and form these islands.41 Furthermore the grafting density of the glycopolymer brushes might be low, hence repulsion between adsorbed side chains is reduced, resulting in the contraction of the backbone.6 Polymer aggregation as a result of strong interactions between the glycopolymer brush and the polar mica substrate could be another reason for only islands forming.3 Furthermore, the polymer was spin-coated from water, and it has been previously reported that an increase in humidity could rearrange polymer molecules to form clusters of several molecules on a mica surface.42
Conclusions
A successful combination of RAFT-mediated polymerisation and ATRP was applied for the synthesis of novel glycopolymer brushes. This work shows the ability of RAFT-mediated polymerisation to control the polymerisation of halogenated monomers for the synthesis of four well-defined ATRP macroinitiators. However, for high target molar mass polymerisations, the molar mass of the macroinitiators deviated from the theoretical molar mass. In addition, an increased PDI was observed in those cases. Radical transfer to halogen in the monomers (ATRP initiator moiety) is thought to be responsible for this limitation,43 which suggests that protective group chemistry on the ATRP initiator moiety is needed in order to obtain well-defined, high(er) molar mass macroinitiators. Details about this work will be reported in a forthcoming publication.
The four ATRP macroinitiators prepared were subsequently used to prepare high molar mass and low PDI glycopolymer brushes with different grafting density along the backbone. This work demonstrated that the Cu(I)Br/n-Pr-1 catalyst system could be successfully used for the polymerisation of unprotected 6-O-MMAGIc in the 'grafting from' process, leading to well-defined glycopolymer brushes. AFM revealed that the molecular brushes adsorbed onto the mica surface, and only islands of polymer molecules were visible, as opposed to the individual brushes reported in earlier studies.7,39
Acknowledgements
The authors greatly acknowledge the Libyan International Centre for Macromolecular Chemistry and Technology for financial support. Novozymes (Pty) Ltd; South Africa is also thanked for their kind donation of Novozym® 435. BK acknowledges support by the South African Research Chair Initiative of the Department of Science and Technology (DST) and the National Research Foundation (NRF) of South Africa.
References
1. Zhang M, Müller AHE. Cylindrical polymer brushes. J Polym Sci Part A: Polym Chem. 2005;43:3461-3481. doi:10.1002/pola.20900 [ Links ]
2. Zhang M, Breiner T, Mori H, Müller AHE. Amphiphilic cylindrical brushes with poly(acrylic acid) core and poly(n-butyl acrylate) shell and narrow length distribution. Polymer. 2003;44(5):1449-1458. doi:10.1016/S0032-3861(02)00774-7 [ Links ]
3. Venkatesh R, Yajjou L, Koning CE, Klumperman B. Novel brush copolymers via controlled radical polymerisation. Macromol Chem Phys. 2004;205:2161-2168. doi:10.1002/macp.200400252 [ Links ]
4. Advincula RC, Brittain WJ, Caster KC, Ruhe J. Polymer brushes: Synthesis, characterisation, applications. Weinheim:Wiley-VCH; 2004. [ Links ]
5. Barbey R, Lavanant L, Paripovic D, et al. Polymer brushes via surface-initiated controlled radical polymerisation: Synthesis, characterisation, properties, and applications. Chem Rev. 2009;109(11):5437-5527. doi:10.1021/cr900045a, PMid:19845393 [ Links ]
6. Lee H, Matyjaszewski K, Yu S, Sheiko S. Molecular brushes with spontaneous gradient by atom transfer radical polymerisation. Macromolecules. 2005;38(20):8264-8271. doi:10.1021/ma051231z [ Links ]
7. Mori H, Müller AHE. New polymeric architectures with (meth)acrylic acid segments. Prog Polym Sci. 2003;28:1403-1439. doi:10.1016/S0079-6700(03)00076-5 [ Links ]
8. Borner HG, Duran D, Matyjaszewski K, Da Silva M, Sheiko S. Synthesis of molecular brushes with gradient in grafting density by atom transfer polymerisation. Macromolecules. 2002;35(9):3387-3394. doi:10.1021/ma012100a [ Links ]
9. Borner HG, Beers K, Matyjaszewski K, Sheiko S, Moller M. Synthesis of molecular brushes with block copolymer side chains using atom transfer radical polymerisation. Macromolecules. 2001;34(13):4375-4383. doi:10.1021/ma010001r [ Links ]
10. Neugebauer D, Sumerlin BS, Matyjaszewski K, Goodhart B, Sheiko S. How dense are cylindrical brushes grafted from a multifunctional macroinitiator. Polymer. 2004;45:8173-8179. doi:10.1016/j.polymer.2004.09.069 [ Links ]
11. Sumerlin BS, Neugebauer D, Matyjaszewski K. Initiation efficiency in the synthesis of molecular brushes by grafting from via atom transfer radical polymerisation. Macromolecules. 2005;38:702-708. doi:10.1021/ma048351b [ Links ]
12. Neugebauer D, Zhang Y, Pakula T, Matyjaszewski K. Heterografted PEO-PnBA brush copolymers. Polymer. 2003;44:6863-6871. doi:10.1016/j. polymer.2003.08.028 [ Links ]
13. Hawker CJ, Bosman AW, Harth E. New polymer synthesis by nitroxide mediated living radical polymerisations. Chem Rev. 2001;101:3661-3688. doi:10.1021/cr990119u, PMid:11740918 [ Links ]
14. Beers KL, Gaynor SG, Matyjaszewski K, Sheiko S, Moller M. The synthesis of densely grafted copolymers by atom transfer radical polymerisation. Macromolecules. 1998;31:9413-9415. doi:10.1021/ma981402i [ Links ]
15. Albertin L, Stenzel MH, Barner-Kowollik C, Foster R, Davis P. Well-defined diblock glycopolymers from RAFT polymerisation in homogeneous aqueous medium. Macromolecules. 2005;38:9075-9084. doi:10.1021/ma051310a [ Links ]
16. You LC, Lu FZ, Li ZC, Zhang W, Li FM. Glucose-sensitive aggregates formed by poly(ethylene oxide)-block-poly(2-glucosyloxyethyl acrylate) with concanavalin A in dilute aqueous medium. Macromolecules. 2003;36:1-4. doi:10.1021/ma025641o [ Links ]
17. Bernard J, Hao X, Davis TP, Barner-Kowollik C, Stenzel MH. Synthesis of various glycopolymer architectures via RAFT polymerisation: From block copolymers to stars. Biomacromolecules. 2006;7:232-238. doi:10.1021/bm0506086, PMid:16398520 [ Links ]
18. Novick SJ, Dordick JS. Preparation of active and stable biocatalytic hydrogels for use in selective transformations. Chem Mater. 1998;10:955-958. doi:10.1021/cm9708123 [ Links ]
19. Palomino E. Carbohydrate handles as natural resources in drug delivery. Adv Drug Deliv Rev. 1994;13:311-323. doi:10.1016/0169-409X(94)90017-5 [ Links ]
20. Wulff G, Zhu L, Schmidt H. Investigations on surface-modified bulk polymers. 1. Copolymers of styrene with a styrene moiety containing a sugar monomer. Macromolecules. 1997;30:4533-4539. doi:10.1021/ma961890z [ Links ]
21. Karamuk E, Mayer J, Wintermantel E, Akaike T. Partially degradable film/fabric composites: Textile scaffolds for liver cell culture. Artif Organs. 1999;23:881-884. doi:10.1046/j.1525-1594.1999.06308.x, PMid:10491038 [ Links ]
22. Okada M. Molecular design and syntheses of glycoploymers. Prog Polym Sci. 2001;26:67-104. doi:10.1016/S0079-6700(00)00038-1 [ Links ]
23. Lowe AB, Sumerlin BS, McCormick CL. The direct polymerisation of 2-methacryloxyethyl glucoside via aqueous reversible addition-fragmentation chain transfer (RAFT) polymerisation. Polymer. 2003;44:6761-6765. doi:10.1016/j.polymer.2003.08.039 [ Links ]
24. Okada M, Tachikawa K, Aoi K. Biodegradable polymers based on renewable resources. II. Synthesis and biodegradability of polyesters containing furan rings. J Polym Sci Part A: Polym Chem. 1997;35:2729-2737. doi:10.1002/(SICI)1099-0518(19970930)35:13<2729::AID-POLA18>3.0.CO;2-D [ Links ]
25. Granville AM, Quémener D, Davis TP, Barner-Kowollik C, Stenzel MH. Chemo-enzymatic synthesis and RAFT polymerisation of 6-methacryloyl mannose: A suitable glycopolymer for binding to the tetrameric lectin concanavalin A. Macromol Symp. 2007;255(1):81-89. doi:10.1002/masy.200750909 [ Links ]
26. Waters Millennium32. Version 3.05. Milford, MA:Waters Corporation. [ Links ]
27. ASTRA® V. Santa Barbara, CA: Wyatt Technology Corporation. [ Links ]
28. Moad G, Chiefari J, Chong BY, et al. Living free radical polymerisation with reversible addition fragmentation chain transfer (the life of RAFT). Polym Int. 2000;49:993-1001. doi:10.1002/1097-0126(200009)49:9<993::AID-PI506>3.0.CO;2-6 [ Links ]
29. Matyjaszewski K, Gaynor SG, Kulfan A, Podwika M. Preparation of hyperbranched polyacrylates by atom transfer radical polymerisation. Macromolecules. 1997;30(17):5192-5194. doi:10.1021/ma970359g [ Links ]
30. Haddleton DM, Crossman MC, Dana BH, et al. Atom transfer polymerisation of methyl methacrylate mediated by alkylpyridylmethanimine type ligands, copper(I) bromide, and alkyl halides in hydrocarbon solution. Macromolecules. 1999;32:2110-2119. doi:10.1021/ma981670g [ Links ]
31. Albertin L, Stenzel M, Barner-Kowollik C, Foster LJR, Davis TP. Well-defined glycopolymers from RAFT polymerisation: Poly(methyl 6-O-methacryloyl-alpha-D-glucoside) and its block copolymer with 2 hydroxyethyl methacrylate. Macromolecules. 2004;37:7530-7537. doi:10.1021/ma049129+ [ Links ]
32. Zhang Y, Huang J, Chen Y. Reactive dendronized copolymer of styryl dendron and maleic anhydride: A single molecular scaffold. Macromolecules. 2005;38(12):5069-5077. doi:10.1021/ma047449n [ Links ]
33. Wang T-L, Lee H-M, Kuo P-L. Functional polymers for colloidal applications. XIV. Syntheses of styrene-maleic anhydride copolymers with different charges and their ability to disperse kaolinite particles. J Appl Polym Sci. 2000;78(3):592-602. doi:10.1002/1097-4628(20001017)78:3<592::AID-APP140>3.0.CO;2-U [ Links ]
34. Saad GR, Morsi RE, Mohammady SZ, Elsabee MZ. Dielectric relaxation of monoesters based poly(styrene-co-maleic anhydride) copolymer. J Polym Res. 2008;15:115-123. doi:10.1007/s10965-007-9150-6 [ Links ]
35. Cheng G, Boker A, Zhang M, Krausch G, Müller AHE. Amphiphilic cylindrical core-shell brushes via a grafting from process using ATRP. Macromolecules. 2001;34(20):6883-6888. doi:10.1021/ma0013962 [ Links ]
36. De Vries A, Klumperman B, De Wet-Roos D, Sanderson RD. The effect of reducing monosaccharides on the atom transfer radical polymerisation of butyl methacrylate. Macromol Chem Phys. 2001;202:1645-1648. doi:10.1002/1521-3935(20010601)202:9<1645::AID-MACP1645>3.0.CO;2-K [ Links ]
37. Russum J, Jones CW, Schork FJ. Continuous reversible addition-fragmentation chain transfer polymerisation in miniemulsion utilizing a multi-tube reaction system. Macromol Rapid Comm. 2004;25:1064-1068. doi:10.1002/marc.200400086 [ Links ]
38. Fleet R, McLeary JB, Grumel V, et al. Preparation of new multiarmed RAFT agents for the mediation of vinyl acetate polymerisation. Macromol Symp. 2007;255(1):8-19. doi:10.1002/masy.200750902 [ Links ]
39. Djalali R, Li S-Y, Schmidt M. Amphipolar core shell cylindrical brushes as templates for the formation of gold clusters and nanowires. Macromolecules. 2002;35(11):4282-4288. doi:10.1021/ma0113733 [ Links ]
40. Sheiko SS, Moller M. Visualization of macromolecules: A first step to manipulation and controlled response. Chem Rev. 2001;101(12):4099-4124. doi:10.1021/cr990129v, PMid:11740928 [ Links ]
41. Muthukrishnan S, Zhang M, Burkhardt M, et al. Molecular sugar sticks: Cylindrical glycopolymer brushes. Macromolecules. 2005;38(19):7926-7934. doi:10.1021/ma0515073 [ Links ]
42. Kumaki J, Hashimoto T. Conformational change in an isolated single synthetic polymer chain on a mica surface observed by atomic force microscopy. J Am Chem Soc. 2003;125(16):4907-4917. doi:10.1021/ja0290429, PMid:12696910 [ Links ]
43. Severac R, Lacroix-Desmazes P, Boutevin B. Reversible addition-fragmentation chain-transfer (RAFT) copolymerisation of vinylidene chloride and methyl acrylate. Polym Int. 2002;51:1117-1122. doi:10.1002/pi.932 [ Links ]
 Correspondence to:
Correspondence to:
Bert Klumperman
Postal address:
Private Bag X1
Matieland 7602, South Africa
email: bklump@sun.ac.za
Received: 30 Aug. 2010
Accepted: 22 Jan. 2011
Published: 22 Mar. 2011
© 2011. The Authors. Licensee: OpenJournals Publishing. This work is licensed under the Creative Commons Attribution License.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}