










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Science
versión On-line ISSN 1996-7489
versión impresa ISSN 0038-2353
S. Afr. j. sci. vol.107 no.3-4 Pretoria mar./abr. 2011
http://dx.doi.org/10.4102/sajs.v107i3/4.459
REVIEW ARTICLE
Gold coordination during homogeneous alkyne and allene cyclisation catalysis: coordination to substrates, to ancillary ligands and in intermediates
Helgard G. RaubenheimerI; Hubert SchmidbaurII
IDepartment of Chemistry and Polymer Science, University of Stellenbosch, South Africa
IIDepartment Chemie, Technische Universität München, Munich, Germany
ABSTRACT
The ever-increasing role of homogeneous gold catalysis in organic synthesis and the consequent need to be able to rationally control the rate and outcome of such reactions has emphasised the importance of each successive metal-carbon coordination step. Concentrating on alkyne and allene cyclisation and upon reaction mechanisms postulated on the basis of empirical and theoretical results, we have examined the coordination of gold fragments to triple bonds, the modification of gold(I) precatalysts to effect specific reaction pathways or enantioselectivity and the isolation of coordinated intermediates or model compounds thereof. Some of the recent advances that have been made in various laboratories are described in this compact review.
Introduction
The observation by W.C. Zeise in 1827 that ethylene forms a complex with a low-valent platinum salt was the key discovery that not only led to the development of one of the most important branches of organometallic chemistry, but also had an enormous economic impact through its relevance to catalysis of organic transformations. Although less well known to the public, and even to many of the scientific community, the catalysis of reactions of unsaturated hydrocarbons, in particular of acetylene, by mercury salts was practised widely after the 1890s and was of great industrial significance. The hydration of acetylene, produced from calcium carbide, to afford acetaldehyde as a key intermediate for many other processes serves as an example.
Although it was known early on that both neighbours of gold in the periodic table - platinum and mercury - were able to complex and activate alkenes and alkynes, it was not until the late 1960s that studies were finally initiated to consider the use of gold salts as catalysts. Progress was slow and there were long periods of inactivity. Now, however, half a century later, homogeneous gold catalysts are in focus of worldwide research. They have become the reagents of choice mainly for a large variety of processes used in the synthesis of high-value fine chemicals, and may soon even enter the production of commodities. Among the other advantages offered, these developments help to enforce a complete banning of mercury in large-scale processes.1
As has been the case with platinum and mercury catalysis, the gold-catalysed reactions have alkenes, allenes and alkynes as substrates, which are complexed and thus activated at the metal centres of gold complexes. In the following analysis of the elementary steps of the association process, alkynes have also been used as the model substrates. Alkynes are also the most important group of compounds transformed by mercury catalysis. Gold-catalysed reactions of alkenes largely follow an analogous pattern.
An early investigation by Berthelot in 1866 showed that the reactions of acetylene (HC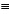 CH, ethyne) with gold salts afforded a brown, insoluble and explosive acetylide, C2Au2, which, when dry, detonated even when touched with a feather. This observation must have discouraged many alchemists and chemists and deterred them from any more detailed studies. Terminal alkynes RCCH also form the corresponding acetylides, which appear as insoluble oligomers [RCCAu]n. This type of reaction, in which the terminal hydrogen atom is substituted by a gold atom, predominates whenever a base is provided to accept the acidic proton. Note that this pronounced affinity of gold cations for HCCH resembles the extreme affinity for the isoelectronic cyanide anion in HCN. In both cases, the chemical bonding between the metal atom and the terminal C-atom, as in Au--CCR and Au--CN, is of the σ-type, based on a highly directional sp-hybrid donor orbital of the carbon atom and a corresponding sdz2 hybrid acceptor orbital of the gold atom, which leads to an end-on bonding associated with a high bond energy and a rigid linear molecular geometry (Chart 1A).2
CH, ethyne) with gold salts afforded a brown, insoluble and explosive acetylide, C2Au2, which, when dry, detonated even when touched with a feather. This observation must have discouraged many alchemists and chemists and deterred them from any more detailed studies. Terminal alkynes RCCH also form the corresponding acetylides, which appear as insoluble oligomers [RCCAu]n. This type of reaction, in which the terminal hydrogen atom is substituted by a gold atom, predominates whenever a base is provided to accept the acidic proton. Note that this pronounced affinity of gold cations for HCCH resembles the extreme affinity for the isoelectronic cyanide anion in HCN. In both cases, the chemical bonding between the metal atom and the terminal C-atom, as in Au--CCR and Au--CN, is of the σ-type, based on a highly directional sp-hybrid donor orbital of the carbon atom and a corresponding sdz2 hybrid acceptor orbital of the gold atom, which leads to an end-on bonding associated with a high bond energy and a rigid linear molecular geometry (Chart 1A).2
By contrast, the affinity of gold salts for internal alkynes RCCR', in which the terminal hybrid orbitals are engaged in bonding of the organic substituents R and R', is much lower, because only the π-orbitals of the triple bond are available for donor-acceptor interactions. This low affinity has made it rather difficult to observe, identify, and finally isolate and characterise any complex formed between these alkynes and common gold salts. The early analytical work relied on elemental analysis and infrared and Raman spectroscopy, which indicated clear stoichiometric ratios and a significant change in the vibrational characteristics of the alkyne substrates. The ν(CC) stretching frequencies were found to be lowered considerably, suggesting a weakening, and hence an activation of the triple bond. As a bonding model, the side-on coordination of a gold atom to the triple bond (π-bonding) was proposed. This approach allowed ligand-to-metal donation and metal-to-ligand back donation into orbitals of appropriate symmetry, following the already widely accepted Dewar-Chatt-Duncanson model, in particular, for the corresponding platinum complexes (Chart 1B).1,2,3
Advanced investigations carried out during the last two decades (1990-2010) have finally provided a plethora of very detailed experimental and theoretical data substantiating the picture of this fundamental interaction.1,3
The chemistry of gold has long shed its older image of being mainly a field of research for curiosity-driven chemists. The booming activity in the area of homogeneous catalysis by well-defined gold compounds has proven the versatility of gold as a single-site catalyst in a wide range of reactions, such as nucleophilic additions to activated alkynes, activation of carbonyls, oxidations, hydrogenations and C-C cross-couplings of the Suzuki type. Many precursor complexes are now commercially available. Not only have significant new methodologies been discovered but their coming of age has been indicated by various applications in total synthesis. Parallel to these developments, many quantum mechanical calculations have been carried out in support of proposed reaction mechanisms, which now also allow predictions for further practical investigations. The tremendous scope of all these developments is attested to by a significant increase in the number of review articles that have been published since 2007.4,5,6,7,8,9,10,11,12,13,14,15,16 Amongst the most productive research groups have been those of Echavarren in Spain, Fürstner and Hashmi in Germany, Toste in the USA and Nolan in the USA, Spain and Scotland.
Based on their research, some general and important conclusions with regard to alkyne cyclisations can be drawn:
1. The cost of gold, seen by many chemists as inhibiting the research and development of its compounds, has ceased to be a factor. More expensive elements like rhodium, palladium and platinum are now used in various commercial catalytic applications. In many instances the ligand cost can be the most expensive component of a chosen catalyst system, making further studies on potential cost-effective systems very important.
2. The development of new catalyst systems is still largely dependent upon trial and error, although certain guidelines regarding privileged structures for various gold-mediated processes have emerged. The capacity for rational design of selective and tunable catalysts for specific and often stereoselective transformations remains a priority.
3. With few exceptions, enantioselective gold catalysis has been achieved using dinuclear gold biphosphine complexes of the general form [(PˆP)(AuX)2] (PˆP = chiral ligand, X = anionic ligand or counterion). However, there is no evidence to support the requirement or participation of two gold centres in the catalytic processes. Examples of enantioselective conversions are still limited and the development of new and useful ligands represents a huge challenge. In all the total syntheses carried out to date, chirality has been retained rather than installed.
4. Cationic, carbophilic gold(I) complexes are most frequently formed by in situ abstraction of halides with silver salts or by preparing linear two-coordinated complexes that contain one labile ligand. In many reactions it is not only the ligands but also the counterions that play a significant, or even a decisive, role. Although Au(III) compounds have been utilised, such precatalysts are more limited and are not discussed in this article.
5. The most common mode of reaction probably involves nucleophilic addition to metal-activated C-C multiple bonds. Most popular are alkyne and allene substrates, and in particular cycloisomerisations of enyne and allene-diene synthons.
6. Although great strides have been made in understanding the kinetics and mechanisms of certain reactions, the search for greater clarity and the identification and description of intermediates needs to be intensified.
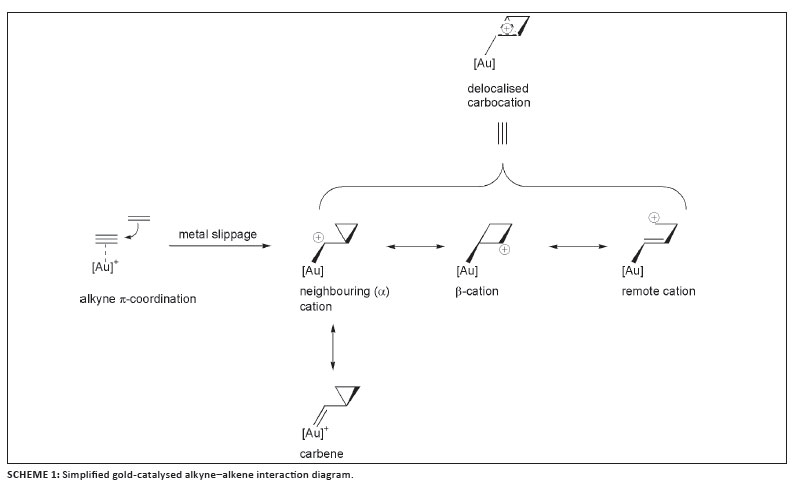
7. The most important intermediates that have been postulated include (vinyl)gold compounds, carbene complexes, delocalised carbocationic complexes and cationic allyl complexes. The extent of carbenoid nature as opposed to simple cationic nature at the carbon adjacent to the metal, or even the delocalised carbocationic functionality, is uncertain; it probably depends on the ligands attached to the metal. A schematic mechanistic scenario introduced by Fürstner for intermediate complex formation during enyne cycloisomerisation (Scheme 1) illustrates the complexity of the bonding in the activated state, and could also serve as a basis for further experimentation.12
8. Owing to relativistic effects, metal-substrate interactions during catalysis are orbitally controlled rather than charge controlled.
This compact review covers the most fruitful advances made in understanding the coordination chemistry involved in cyclisation catalysis of alkynes and allenes by gold compounds since 2008, and includes literature up to August 2010. Older ligand modifications for the precatalysts have been reviewed by Toste and co-workers (Gorin et al.8).
Stoichiometry and structure of (alkyne)gold complexes
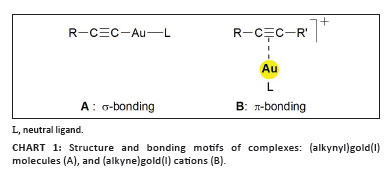
Presently, an inventory of the chemistry of (alkyne)gold complexes shows that the vast majority of compounds have alkynes coordinated to gold(I) centres, while the corresponding compounds of gold(II) and gold(III) are rare and poorly characterised. The gold(I) complexes are either neutral or cationic systems in which the alkyne is attached to neutral AuX or to cationic (L)Au+ or (L^L)Au+ units. These units differ in their acceptor strength depending on the nature of the anions X- and ligands L, and even the counterions Y- associated with the (L)Au+ cations or their solvates may play a role in the affinity towards the alkyne substrates. In standard cases, the gold atom is linearly two-coordinated with the alkyne occupying one of the two trans acceptor sites on the metal atom, but examples with bidentate anions L^X- and neutral ligands L^L are also known, where the coordination number of the metal atom increases from 2 to 3. Complexes with two alkynes attached to the same gold atom have not been fully characterised but, very recently, examples have been discovered in which an alkyne triple bond accepts two (L)Au+ units. In these cations, obviously both π-orbitals are engaged in ligand-to-metal bonding, but this mode of interaction appears only if metal acetylides are involved as substrates (Chart 2).1
Structural work on several prototypes of the general formula (alkyne)AuCl has confirmed the proposed π-complexation ('side-on') of the alkyne. For symmetrical alkynes like EtCCEt and cyclo-dodecyne, the gold atom is equidistant from the two triple-bonded carbon atoms, with average Au-C distances of 2.16 Å. Upon complexation, the CC distance is lengthened considerably, from about 1.20 Å in the free alkynes to about 1.235 Å in the adducts. Another striking observation relates to the distortion of the alkynes from linearity. The CC-C angles are bent from 180º in the strain-free alkynes to about 165º in the direction away from the approaching metal atom. As expected, these deviations are even larger for compounds with a bidentate anionic ligand, LˆX- (e.g. a triaza-pentadienide) where the gold atom has become tricoordinate, and unsymmetrical substitution of the alkyne as in RCCR' leads to a slight shift of the gold atom to the more electron-rich and less sterically demanding substituent (Chart 3).17,18

This very significant structural evidence is complemented by the considerable shift of the ν(CC) frequency to lower wave numbers (already mentioned above). The differences Δν can amount to 350 cm-1. In the nuclear magnetic resonance (NMR) spectra of the complexes, the chemical shifts of the 1H and 13C nuclei are not much affected, probably owing to a cancellation of opposing parameters, and 197Au Mössbauer spectroscopy has confirmed that the oxidation state of the metal atom is unchanged in the complexes.1
There are no experimental data yet defining the enthalpy of formation of the complexes from their components, but several state-of-the-art theoretical calculations of model gold-alkene/alkyne compounds with C2v symmetry in the gas phase have indicated that binding energies are always higher for C=C bonds than for CC ones. From this result it follows that the selective alkynophilicity observed in homogeneous catalysis for systems that contain alkene and alkyne functions is not directly related to a thermodynamic preference during the initial coordination step but has a kinetic origin.19,20
The general bonding pattern is largely the same in cationic complexes with an auxiliary ligand, of the type [(L)Au(alkyne]+, with L representing, for example, a tertiary phosphine R3P or an N-heterocyclic carbene (NHC).18,21 In practice, solutions containing these cations are prepared by treating complexes [(L)AuCl] with silver salts with weakly coordinating anions, such as AgBF4, AgPF6, AgSbF6 or AgOSO2CF3 (AgOTf), in dichloromethane, acetonitrile or other solvents, followed by addition of the alkyne. Precursor complexes with X representing a bis(sulfonyl)imid ligand, as in [(L)AuN(SO2R)2], react directly with alkynes to give solutions of the corresponding cationic complexes. These preparations have the advantage that further studies can be carried out in the absence of silver salts, which may have an influence on subsequent reactions (Chart 3).22,23
In attempts to isolate compounds of the type [(L)Au(alkyne)]+ Y- for further characterisation, the bulky and electron-rich tri(tert-butyl)phosphine ligand t-Bu3P proved to be particularly useful. Crystallisation of [(t-Bu3P)Au(MeCCt-Bu)]+ SbF6- has recently provided a set of important complementary data. Moreover, it was in this work that it could be demonstrated that alkynes can accommodate two [(t-Bu3P)Au]+ cations, employing both π-orbitals in their perpendicular orientation.24
Recent theoretical studies have shown that the (alkyne)--MI interaction is based on both efficient alkyne σ-donation and equally important π-acceptance, which are both stronger for M = Au than for M = Cu, Ag. However, contrary to the robust σ-donation, the π-back donation is more sensitive to the nature of the auxiliary ligands L or X- in cationic or neutral complexes, respectively, and to structural restraints. Obviously, both factors need to be fine-tuned in attempting to tailor the performance of a catalyst, which in a crucial first step activates the alkyne by the π-complexation described above.3
As already mentioned above, no such detailed information is as yet available for alkyne complexes of gold(II) and gold(III), even though numerous transformations of alkynes have been successfully catalysed by salts and complexes with gold in these higher oxidation states.1
New and selective catalysts
Although a large number of precatalysts used belong to known complex types, completely new gold complexes have also been introduced and utilised.
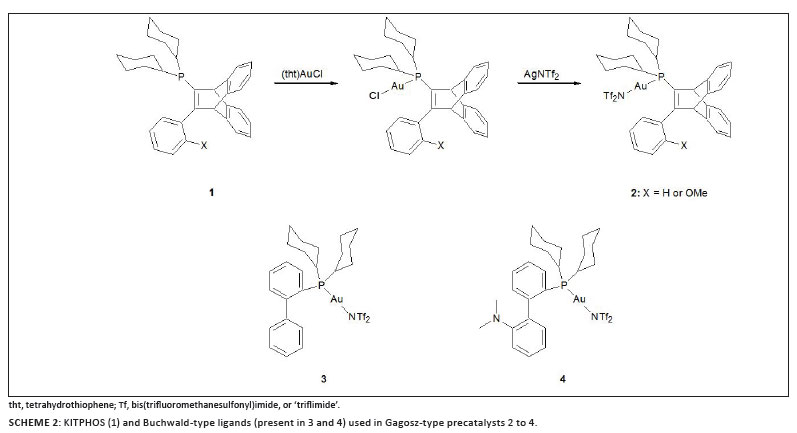
Employing a screening test for potential catalysts that included four different cycloisomerisations, i.e. the cyclisation of w-alkynylfurans, the cyclisation of 2-alkynylaryl epoxides and N-propargylcarboximides, as well as intramolecular hydroxylation and hydroxycarboxylation, Hashmi and co-workers compared the activity of KITPHOS (11-dicyclohexylphosphino-12-phenyl-9,1-ethenoanthracene derivatives) (1) and Buchwald-type (3,4) phosphine ligands when attached to gold(I) (Scheme 2).25 The synthetic procedure followed was first described by Mezailles et al.23 and afforded triflimide (NTf2) complexes, circumventing the need for further removal of chloride by silver salts during the activation process in catalysis, as already pointed out. Although the KITPHOS complexes are generally highly efficient catalysts for a range of cyclisations, they do not consistently perform better than their Buchwald analogues.
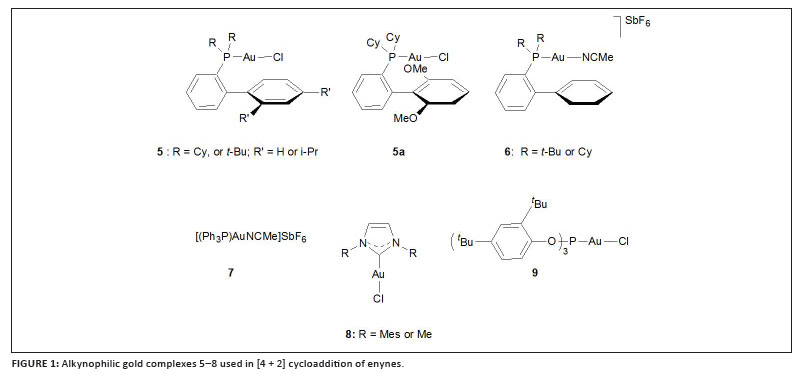
In order to carefully investigate the [4 + 2] cycloadditions of alkenyl- and aryl-substituted 1,6-enynes, highly alkynophilic Au(I) complexes are required. For this purpose Echavarran and co-workers prepared the series of gold complexes shown in Figure 1.26 The new cationic phosphite complex generated from 9, particularly, is most active and cyclisations occur rapidly under mild conditions. The mechanism of the reaction was studied using density functional theory (DFT) calculations and deuterium labelling. Intermediates that form during the cycloaddition of arylalkynes include a gold carbene complex, a (vinyl)gold carbocation and a neutral (vinyl)gold complex. The aryl substituent is believed to stabilise both of the former two intermediates by π-interaction.
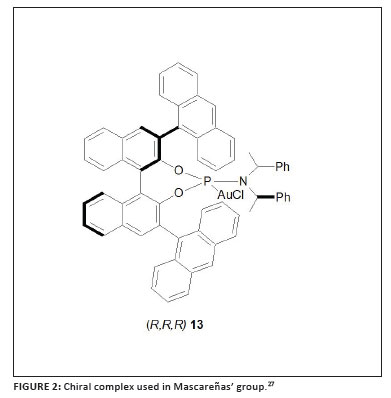
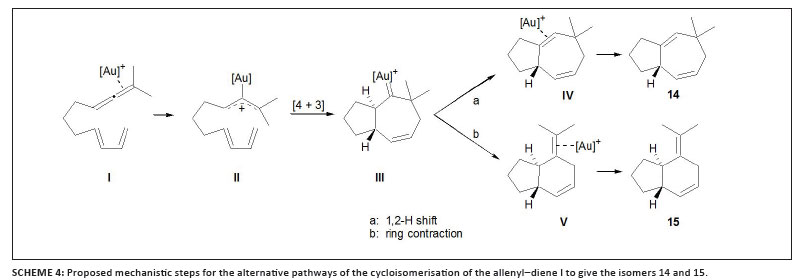
A series of investigations, all involving selective [4 + 2] and/or [4 + 3] cycloadditions within allene-dienes, superbly illustrate the role of ligand control in such catalysed conversions. Using the well-known NHC gold complex [(IPr)AuCl] (IPr = N,N'-bis(2,6-diisopropylphenyl) imidazol-2-ylidene, compare 8 with R = i-Pr) in combination with AgSbF6 as catalyst, Mascareñas and co-workers established that the allene-diene model substrate 10 in Scheme 3 is converted mainly into the [4 + 3] adduct 11 although a [4 + 2] cycloadduct, 12, is also formed.27 Then Toste and co-workers and Mascareñas and co-workers independently reported selective syntheses, while reasoning along similar lines, and successfully manipulated the neutral ligands attached to gold.28,29 Having postulated a carbocationic intermediate for the [4 + 2] pathway and a cationic gold carbenoid complex in the [4 + 3] cycloaddition, Toste and co-workers selectively prepared the latter product by using the electron-rich, and thus strong σ-donor ligand, di(t-butyl)phenyl phosphine.28 On the other hand, a typical π-acceptor ligand like (PhO)3P attached to gold diverts the model reaction as well as other related transformations to [4 + 2] cycloadduct formation by presumably stabilising the carbocationic intermediate. Mascareñas and co-workers, after having established a high chemoselectivity and complete stereoselectivity in the [4 + 2] cycloaddition while using the bulky phosphite ligand in 9 (Figure 1), took another step forward. They demonstrated the first examples of effective asymmetric catalysis by a monomeric, carbophilic P-donor gold complex using chiral phosphoramidite-based cationic gold complexes to provide the targeted [4 + 2] product enantioselectively and in high yield.29 The most successful precatalyst has been the complex (R,R,R)13, shown in Figure 2.
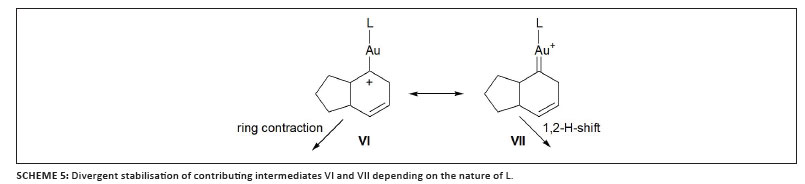
Based on the results of extensive mechanistic investigations, supported by DFT calculations for model systems, the authors eventually postulated a common pathway for both [4 + 3] and [4 + 2] cycloadditions that includes the consecutive formation of cationic gold allyl (II) and cycloheptenyl gold carbene (III) complexes (Scheme 4). Respective 1,2-H shift or ring contraction finally leads to the two product types 14 or 15. This result was independently confirmed by Toste and co-workers, who also emphasised the important role of steric influences in the catalysed reactions.30 Later, Fürstner interpreted a similar ring-contraction fostered by strong π-acidic ligands to be as a result of the presence of a largely carbocationic centre attached to gold (VI), whereas greater typical carbene character (VII) by virtue of better donating ancillary ligands would result in a preferred 1,2-H shift (discussed above).31
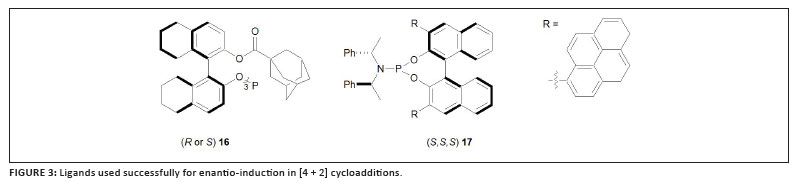
Toste and co-workers also reported on enantio-induction in [4 + 2] cycloadditions of allene-dienes with linear, cationic gold complexes that bear strong π-acceptor ligands.32 The C3-symmetric phosphite complexes with the ligands (R or S)16 (Figure 3) have been the most successful for the preparation of trans-hexahydroindenes and complexes with (S,S,S)17, an ortho-arylphosphoramidite, to realise pyrrolidine products. Again, as was also established by Mascareñas, ee% yields in these reactions are sometimes significantly influenced by the counterions used.29
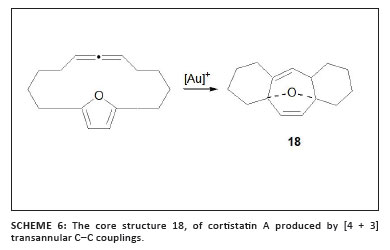
Gung et al.33 applied the empirical results discussed above in an attempt to force a [4 + 3] cycloaddition and produce the core structure of the natural product cortistatin A (18 in Scheme 6) while using a macrocyclic allene substrate. Neither satisfactory yields nor selectivity responses were found with a series of electron-rich and electron-poor gold complexes that contained phosphine or phosphite ligands. However, exclusive [4 + 3] transannular cycloaddition occurs when macrocyclic propargyl acetates are converted with the electron-rich ligand biphenyl(t-Bu)P coordinated to gold(I).
When changing from P-donor ligands to NHCs, Fürstner showed that, whereas the π-acceptor properties of the latter ligands are often considered to have little impact, their valence π-type orbitals, and thus their π-acidic properties, are more easily tuned than their σ-donor characteristics.31Furthermore, when these ligands are attached to gold, such effects can be exploited to influence the course of different processes. The ligands shown in Table 1 have been prepared and their HOMO (Eσ) and LUMO (Eπ) energies calculated using density functional theory. Electrochemical potentials of their corresponding rhodium(I) complexes of the type [(L)RhCl(cod)] have also been determined.
Clearly, the attachment of cyclophanic rings to a carbene ligand like 19 imparts significant changes in the ligating properties of 20-22, and the π-type orbitals are more responsive than the σ-donor orbitals. The bonding properties of the ligands concur with the redox potentials of the corresponding rhodium complexes. Ligand 21 exerts by far the best and 19 the weakest π-acceptor properties. In complexes 20-22, differences are governed by strong but opposing interactions between the two aromatic planes in each cyclophane skeleton. Introducing a triazo ring in 23 reduces the electron donor capacity, whereas conversion of the five-membered ring in 19 into an ylid 24 produces the strongest carbene donor of all. The following results illustrate the effective modification of the π-acceptor properties of the auxiliary ligand. A cationic gold complex of the strong σ-donor ligand 19 exclusively produces the [4 + 3] adduct in the cyclisation of an allene-diene, whereas the [4 + 2] pathway is favoured with the electron-poorer combination 20.AuCl/AgBF4, in accordance with the interpretation given above (Scheme 5). Completely different behaviour exhibited by the same two complexes is also seen during the cycloaddition of ene-allenes where exclusive discrimination between [3 + 2] and [2 + 2] adduct formation occurs.
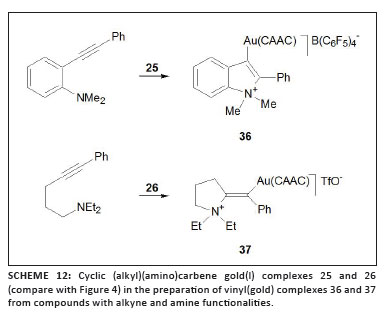
Bertrand and co-workers introduced cyclic (alkyl)(amino)carbene (CAAC) ligands into gold chemistry and utilised such gold complexes in the intermolecular hydroamination of alkynes.34 Employing the two gold catalysts 25 (in combination with AgOTf) and 26 (Figure 4), they discovered the promotion of unusual hydroammoniumation and methylamination of various N-methyl-2-(2-organoethynyl)anilines by carboamination of the alkyne function. Intermediates formed during the course of these reactions could be isolated (and are described below).35
Echavarren and co-workers have been responsible for a great resurgence of the chemistry of the first ever class of carbene complexes36 prepared by Bonati and co-workers nearly 40 years ago.37 A series of hydrogen bond supported heterocyclic carbenes (HBHCs), derived from 2-pyridyl isocyanide (Chart 4), were shown to be active catalysts in reactions of 1,6-enynes. When comparing these to gold complexes bearing noncyclic carbene ligands derived from 4-pyridyl isocyanide (D and E), significantly different results were obtained that warrant mentioning:
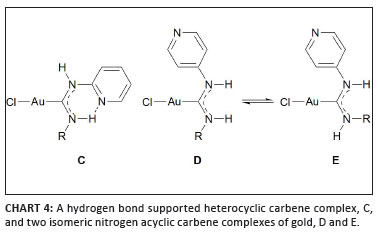
• Effective catalysts to produce dienes by skeletal rearrangement contain the 2-pyridyl group.
• Catalytic activity of the catalyst that contains a 4-pyridyl group is very low, presumably reflecting intermolecular blocking of the active Au(I) centre by N-coordination.
• Whereas previously mentioned complexes like [(IPr)AuNCPh]+ favour exo-cyclisation, endo products are produced by certain HBHCs.
• Extremely high activity is found in related methoxycyclisations that have been studied in methanol; here, all carbenes attached to gold are open.
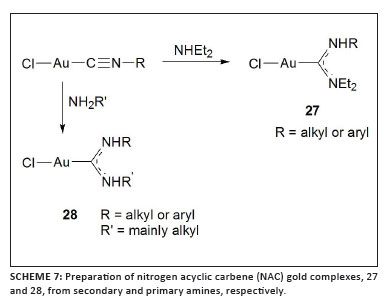
Inspired in particular by the final result mentioned above, Echavarren and co-workers then studied the skeletal rearrangement in other cyclisations of 1,6-enynes employing 12 nitrogen acyclic carbene (NAC) gold complexes (27 and 28 in Scheme 7) prepared by reaction of a range of isocyanogold complexes with diethylamine or various primary amines.38 Up to three different cyclisation products were obtained from a model enyne and the product ratios depended upon the substituents on the carbene. In general, selectivity can be high and the (NAC)gold(I) complexes are at least as reactive and selective as previously reported more elaborate carbene complexes of gold(I), and the yields and rates of conversions are higher than those achieved with comparable (HBHC)Au(I) catalysts. Finally, the nucleophilic addition of dibenzoylmethane to a 1,6-enyne was investigated to determine the electrophilicity of these ligands. Highly electrophilic catalysts bearing, for example, a bulky phosphine ligand preferentially afforded the product 29 (Scheme 8) as a result of stabilisation of the cationic character of the intermediate. All the NAC ligands studied reflected a highly donating character that is instrumental in preferentially facilitating the reaction via a carbenoic intermediate to form 30.
Several isonitrile gold complexes were also converted to (NAC)gold complexes by Hashmi and co-workers.39 They reported 12 crystal structures and results of reactivity studies of the complexes in a phenol synthesis in which furanyn was used as a substrate. Extremely high turnover numbers were found, indicating that these more classic complexes outperform (KITPHOS)gold(I) complexes, (NHC)gold(I) complexes and Buchwald-ligand complexes of gold under similar reaction conditions. It is noteworthy that a broad range of activities were found when the counterions were varied by the addition of AgX (X = SbF5, PF6, OTf, OTs, BF4, NTf2 and ONf) to the NAC gold chloride complexes in the catalyst mixture.
Intermediate coordination
Vinyl complexes of gold are assumed to form transiently during the activation and subsequent nucleophilic attack of alkynes and allenes, as illustrated in simplified form in Scheme 1. After Hashmi and co-workers40 prepared a number of (vinyl)gold complexes by following the boronic acid route developed by Partyka et al.41, namely

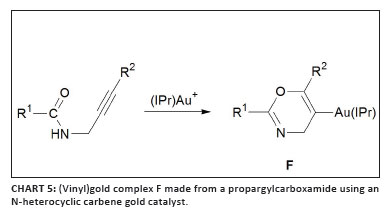
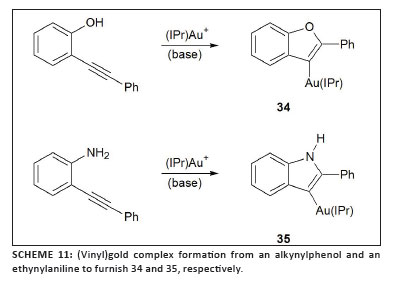
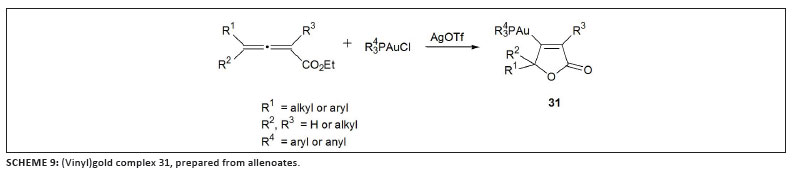
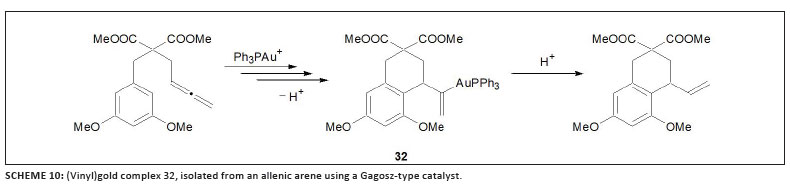
three groups published other examples of such complexes in 2009. Liu and Hammond42 studied the cyclisation of allenoates with trialkylphosphine or triarylphosphine gold complexes in stoichiometric quantities and prepared stable complexes of type 31 (Scheme 9). Such compounds were then successfully used as transmetallation reagents in palladium-catalysed cross-coupling.43 While investigating the 'seemingly simple' catalytic cycle for the cycloisomerisation of allenic arenes employing the Gagosz catalyst, [Ph3PAuNTf2] (Scheme 10),23 Gagné and co-workers isolated the intermediate (vinyl)gold complex 32.44 Furthermore, they established that under catalytic conditions the catalyst rests as another intermediate, 33 (Figure 5). Earlier, theoretical results obtained by the research group of Toste and Morganelli indicated that such species are involved in the cycloisomerisation of enynes.45 Subsequently, Hashmi et al.46 developed a method to isolate vinylgold species from propargylcarboxamides that contain an internal alkyne group using the cationic gold fragment [(IPr)Au]+ (F, Chart 5). The scope of this approach could be broadened to permit (vinyl)gold formation by 5-endo-dig cyclisation from an ortho-alkynylphenol and 2-(phenylethynyl)aniline,47 affording complexes 34 and 35 (Scheme 11).

Using (CAAC)gold(I) complexes (see above and Figure 4), Bertrand and co-workers carried out related cyclisation reactions in attempts to isolate previously postulated three-coordinated gold intermediates.35 Two cationic (vinyl)gold complexes 36 and 37 were successfully isolated and authenticated (Scheme 12).
Although they are often postulated as intermediates in homogeneous gold catalysis, very few gold carbene complexes that are not stabilised by one or two adjacent heteroatoms have been isolated and described in detail.48,49 The formation of a free cationic carbene complex,
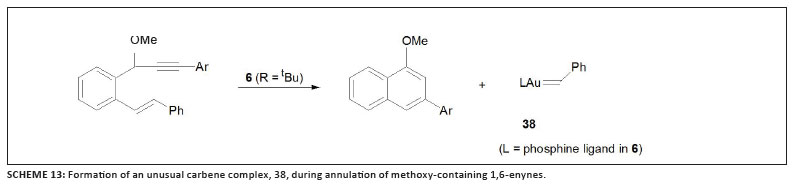
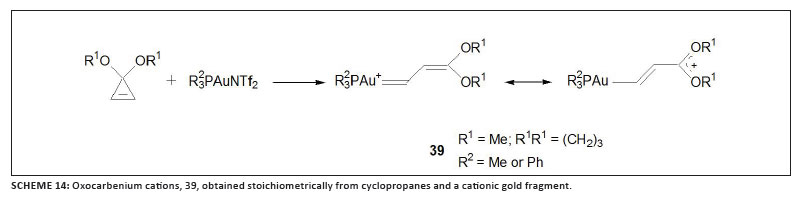
[(L)Au=CHPh]+, 38 (featuring the same ligand L as in catalyst 6 with R = t-Bu), during the annulation of 1,6-enynes that carry OMe substituents at the propargyl position (Scheme 13), has been disclosed, and convincingly verified.50 The formation of the carbene complex is a consequence of a rare retro-cyclopropanation. Following a well-established route to prepare various carbene complexes of metals, including the Grubbs catalyst,51,52 Fürstner and co-workers combined substituted cyclopropanes with a labile gold complex as shown in Scheme 14.53 Both products obtained can best be described as thermally interconvertible geometric isomers of oxocarbenium cations with C=C double bonds located a to the gold atom and with their positive charges stabilised by two oxygen atoms.
Note that the intermediate oxonium compound that was identified during the stoichiometric Hammond synthesis of gold-vinyl complexes42 is related to Fürstner's cationic compounds. Although not specifically mentioned by the former authors, both their intermediate compound and the various products can also be represented by carbene complex mesomeric structures (Scheme 15). The relatively high 13C NMR chemical shift (d 205) for the coordinated carbon atom in the one fully described neutral product concurs with at least some carbocationic carbene character.54 Unfortunately, the corresponding value for the intermediate has not been reported. Such ligated carbons in Fürstner's compounds resonate at d values > 210. This result should not eclipse the fact that other experimental and physical data for these cationic compounds are fully compatible with those of gold-vinyl structures, as discussed above.
After metal slippage55,56 and nucleophilic attack during cycloisomerisation of enynes (Scheme 1) the issue that emerges57,58 is not whether so-called carbene complexes are formed. Aminocarbene complexes of the Fischer-type also carry significant positive charges on their nitrogen atoms and their C-N bonds exhibit significant barriers of rotation suggesting preponderance of a zwitterionic state, and still they are generally classified as carbene complexes.
Questions of both a more fundamental and a practical nature remain:
1. To what extent should metal-carbon double bond character be invoked when explaining reactivity and transition or intermediate state stabilisation or, alternatively, to what extent is the cationic charge delocalised?
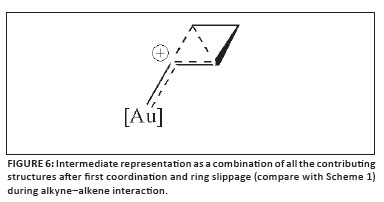
2. How could the different mesomeric contributions that constitute the delocalised representation in Figure 6 (compare again Scheme 1) be logically and selectively enhanced, and expressed, by the tailoring of ancillary ligands to allow discrimination between different available catalysed pathways while considering different modes of activation? As we have reported, important progress has already been made in this respect.
3. Could suitable model gold complexes without heteroatoms in their alkyne-derived ligands or analogues thereof be prepared and studied in order to establish the contribution (or activity) of different canonical (or tautomeric) structures?
Protodeactivation plays a key role in gold-mediated catalytic processes and catalyst regeneration. Experiments to determine the relative kinetic basicities of a series of neutral gold compounds were conducted by Roth and Blum.59 The gold-coordinated carbon basicities conform to the trend sp3 < sp < sp2 in (Ph3P)AuX with X equal to CH3 < t-BuCC < Ph < vinyl, which is different from the normal basicity trend of the related anions based on their pKb values (sp << sp2< sp3).60 Care should however be exercised in drawing such comparisons because of the important role that the solvent might play. Notably, precatalysts bearing the NHC ligand IPr, protodeaurate nearly twice as effectively as their phosphine analogues. Despite the unexplained results obtained for some heteroatom-containing (vinyl)gold compounds, these findings offer useful information applicable to the future design of catalysts.
Conclusions
Confining ourselves mostly to intramolecular addition reactions catalysed by gold(I) compounds, impressive developments in clarifying the consecutive changes in gold coordination during the total process have been described. Many more opportunities and challenges exist and these have to be addressed before the outcome of reactions through tuning of auxiliary ligands can be predicted with confidence. Total manipulation of such reactions will only be possible if the reaction mechanisms and, hence, the important intermediates that form are known, and their metal-ligand bonding is understood on a fundamental level. The presently developed frameworks and hypotheses should be rigorously challenged experimentally and theoretically in attempts to alter and refine them. Meticulous kinetic investigations should be a priority.
Acknowledgements
Financial support (to HGR) from the Oppenheimer Memorial Fund is gratefully acknowledged.
References
1. Schmidbaur H, Schier A. Gold η2-coordination to unsaturated and aromatic hydrocarbons: The key step in gold-catalyzed organic transformations. Organometallics. 2010;29:3-27. doi:10.1021/om900900u [ Links ]
2. Schmidbaur H, Schier A. Organogold chemistry. In: Crabtree RH, Mingos DMP, editors. Comprehensive organometallic chemistry III. Vol. 2. Oxford: Elsevier, 2006; p. 251-308. [ Links ]
3. Salvi N, Belpassi L, Tarantelli F. On the Dewar-Chatt-Duncanson model for catalytic gold(I) complexes. Chem Eur J. 2010;16:7231-7239. PMid:20468042 [ Links ]
4. Gorin DJ, Toste FD. Relativistic effects in homogeneous gold catalysis. Nature. 2007;446:395-403. doi:10.1038/nature05592, PMid:17377576 [ Links ]
5. Hashmi ASK. Gold catalyzed organic reactions. Chem Rev. 2007;107:3180-3211. doi:10.1021/cr000436x, PMid:17580975 [ Links ]
6. Arcadi A. Alternative synthetic methods through new developments in catalysis by gold. Chem Rev. 2008;108:3266-3325. doi:10.1021/cr068435d, PMid:18651778 [ Links ]
7. Li Z, Brouwer C, He C. Gold catalyzed organic transformations. Chem Rev. 2008;108:3239-3265. doi:10.1021/cr068434l, PMid:18613729 [ Links ]
8. Gorin DJ, Sherry BD, Toste FD. Ligand effects in homogeneous Au catalysis. Chem Rev. 2008;108:3351-3378. doi:10.1021/cr068430g, PMid:18652511, PMCid:2754803 [ Links ]
9. Jiménez-Núñez E, Echavarren AM. Gold-catalyzed cycloisomerizations of enynes: A mechanistic perspective. Chem Rev. 2008;108:3326-3350. doi:10.1021/cr0684319, PMid:18636778 [ Links ]
10. Widenhoefer RA. Recent developments in enantioselective gold(I) catalysis. Chem Eur J. 2008;14:5382-5391. doi:10.1002/chem.200800219, PMid:18442031 [ Links ]
11. Belmont P, Parker E. Silver and gold catalysis for cycloisomerization reactions. Eur J Org Chem. 2009;6075-6089. doi:10.1002/ejoc.200900790 [ Links ]
12. Fürstner A. Gold and platinum catalysis - a convenient tool for generating molecular complexity. Chem Soc Rev. 2009;38:3208-3221. doi:10.1039/b816696j, PMid:19847352 [ Links ]
13. Nevado C. Gold catalysis: Recent developments and future trends. CHIMIA. 2010;64:247-251. doi:10.2533/chimia.2010.247 [ Links ]
14. Sengupta S, Shi X. Recent advances in asymmetric gold catalysis. ChemCatChem. 2010;2:609-619. doi:10.1002/cctc.201000070 [ Links ]
15. Garcia P, Malacria M, Aubert C, Gandon V, Fensterbank L. Gold-catalysed cross-couplings: New opportunities for C-C bond formation. ChemCatChem. 2010;2:493-497. doi:10.1002/cctc.200900319 [ Links ]
16. De Mendoza P, Echavarren AM. Synthesis of arenes and heteroarenes by hydroarylation reactions catalyzed by electrophilic metal complexes. Pure Appl Chem. 2010;82:801-820. doi:10.1351/PAC-CON-09-10-06 [ Links ]
17. Wu J, Kroll P, Raska Dias HV. Gold(I) chloride coordinated 3-hexyne. Inorg Chem. 2009;48:423-425. doi:10.1021/ic8020854, PMid:19072612 [ Links ]
18. Flügge S, Anoop A, Goddard R, Thiel W, Fürstner A. Structure and bonding in neutral and cationic 14-electron gold alkyne π-complexes. Chem Eur J. 2009;15:8558-8565. doi:10.1002/chem.200901062, PMid:19569141 [ Links ]
19. Nechaev MS, Rayon VM, Frenking G. Energy partitioning analysis of the bonding in ethylene and acetylene complexes of group 6, 8 and 11 metals. J Phys Chem A. 2004;108:3134-3142. doi:10.1021/jp031185+ [ Links ]
20. Garcia-Mota M, Cabello N, Maseras F, Echavarren M, Perez-Ramirez J, Lopez N. Selective homogeneous and heterogeneous gold catalysis with alkynes and alkenes: Similar behaviour and different origin. ChemPhysChem. 2008;9:1624-1629. doi:10.1002/cphc.200800246, PMid:18537219 [ Links ]
21. Akana J, Bhattacharyya KX, Müller P, Sadighi J. Reversible C-F bond formation and the Au-catalyzed hydrofluorination of alkynes. J Am Chem Soc. 2007;129:7736-7737. doi:10.1021/ja0723784, PMid:17547409 [ Links ]
22. Gagosz F. Recent developments in gold catalysis. Tetrahedron. 2009;65:1757-1767. doi:10.1016/j.tet.2008.12.040 [ Links ]
23. Mezailles N, Ricard L, Gagosz F. Phosphine gold(I) bis(trifluoromethanesulfonyl)-imidate complexes as highly efficient and air-stable catalysts for the cycloisomerization of enynes. Org Lett. 2005;7:4133-4136. doi:10.1021/ol0515917, PMid:16146370 [ Links ]
24. Hooper TN, Green M, Russell CA. Cationic Au(I) complexes: Synthesis, structure and reactivity. Chem Commun. 2010;46:2313-2315. doi:10.1039/b923900f, PMid:20234943 [ Links ]
25. Hashmi SAK, Loos A, Littmann A, et al. Gold(I) complexes of KITPHOS monophosphines: Efficient cycloisomerization catalysts. Adv Synth Catal. 2009;351:576-582. doi:10.1002/adsc.200800681 [ Links ]
26. Nieto-Oberhuber C, Pérez-Galán P, Herrero-Gómez E, et al. Gold(I)-catalyzed intramolecular [4 + 2] cycloadditions of arylalkynes or 1,3-enynes with alkenes: Scope and mechanism. J Am Chem Soc. 2008;130:269-279. doi:10.1021/ja075794x, PMid:18076170 [ Links ]
27. Trillo B, López F, Montserrat S, et al. Gold-catalyzed [4C + 3C] intramolecular cycloaddition of allene-dienes: Synthetic protocol and mechanistic implications. Chem Eur J. 2009;15:3336-3339. doi:10.1002/chem.200900164, PMid:19229922 [ Links ]
28. Mauléon P, Zeldin RM, González AZ, Toste FD. Ligand-controlled access to [4 + 2] and [4 + 3] cycloadditions in gold-catalyzed reactions of allene-dienes. J Am Chem Soc. 2009;131:6348-6349. doi:10.1021/ja901649s, PMid:19378998, PMCid:2711638 [ Links ]
29. Alonso I, Trillo B, López F, et al. Gold catalyzed [4C + 2C] cycloadditions of allenedienes, including an enantioselective version with new phosphoramidite-based catalysts: Mechanistic aspects of the divergence between [4C + 3C] and [4C + 2C] pathways. J Am Chem Soc. 2009;131:13020-13030. doi:10.1021/ja905415r, PMid:19697936 [ Links ]
30. Benitez D, Tkatchouk E, González AZ, Goddard (III) WA, Toste FD. On the impact of steric and electronic properties of ligands on gold(I)-catalyzed cycloaddition reactions. Org Lett. 2009;11:4798-4801. doi:10.1021/ol9018002, PMid:19780543, PMCid:2783583 [ Links ]
31. Alcarazo M, Stork S, Anoop A, Thiel W, Fürstner A. Steering the surprisingly modular π-acceptor properties of N-heterocyclic carbenes: Implications for gold catalysis. Angew Chem Int Ed. 2010;49:2542-2546. doi:10.1002/anie.200907194, PMid:20209549 [ Links ]
32. González A, Toste FD. Gold(I)-catalyzed enantioselective [4 + 2]-cycloaddition of allene-dienes. Org Lett. 2010;12:200-203. doi:10.1021/ol902622b, PMid:19961192, PMCid:2798906 [ Links ]
33. Gung BW, Craft DT, Bailey LN, Kirschbaum K. Gold-catalyzed transannular [4 + 3] cycloaddition reactions. Chem Eur J. 2010;16:639-644. doi:10.1002/chem.200902185, PMid:19937623 [ Links ]
34. Zeng X, Frey GD, Kinjo R, Donnadieu B, Bertrand G. Synthesis of a simplified version of stable, bulky and rigid cyclic alkyl(amino) carbenes and catalytic activity of ensuing gold(I) complex in the three-component preparation of 1,2-dihydroquinoline derivatives. J Am Chem Soc. 2009;131:8690-8696. doi:10.1021/ja902051m, PMid:19456108, PMCid:2724870 [ Links ]
35. Zeng X, Kinjo R, Donnadieu B, Bertrand G. Serendipitous discovery of the catalytic hydroammoniumation and methylamination of alkynes. Angew Chem Int Ed. 2010;49:942-945. [ Links ]
36. Bartholomé C, Ramiro Z, Pérez-Galán P, et al. Gold(I) complexes with hydrogen-bond supported heterocyclic carbenes as active catalysts in reactions of 1,6-enynes. Inorg Chem. 2008;47:11391-11397. doi:10.1021/ic801446v, PMid:18947178 [ Links ]
37. Minghetti G, Bonati F. Bis(carbene) complexes of gold(I) and gold(III). J Organomet Chem. 1973;54:C62-C63. [ Links ]
38. Bartolomé C, Ramiro Z, Garcia-Cuadrado D, et al. Nitrogen acyclic gold(I) carbenes: Excellent and easily accessible catalysts in reactions of 1,6-enynes. Organometallics. 2010;29:951-956. doi:10.1021/om901026m [ Links ]
39. Hashmi SAK, Hengst T, Lothschütz C, Rominger F. New and easily accessible nitrogen acyclic gold(I) carbenes: Structure and application in the gold-catalyzed phenol synthesis as well as the hydration of alkynes. Adv Synth Catal. 2010;352:1315-1337. doi:10.1002/adsc.201000126 [ Links ]
40. Hashmi SAK, Ramamurthi TD, Rominger F. Synthesis, structure and reactivity of organogold compounds of relevance to homogeneous gold catalysis. J Organomet Chem. 2009;694:592-597. doi:10.1016/j. jorganchem.2008.11.054 [ Links ]
41. Partyka DV, Zeller M, Hunter AD, Gray TG. Relativistic functional groups: Aryl carbon-gold bond formation by selective transmetalation of boronic acids. Angew Chem Int Ed. 2006;45:8188-8191. doi:10.1002/anie.200603350, PMid:17111449 [ Links ]
42. Liu L-P, Hammond GB. Reactions of cationic gold(I) with allenoates: Synthesis of stable organogold(I) complexes and mechanistic investigations on gold-catalyzed cyclizations. Chem Asian J. 2009;4:1230-1236. doi:10.1002/asia.200900091, PMid:19472292 [ Links ]
43. Hashmi ASK, Lothschütz C, Döpp R, Rudolph M, Ramamarthi TD, Rominger F. Gold and palladium combined for cross-coupling. Angew Chem Int Ed. 2009;48:8243-8246. doi:10.1002/anie.200902942, PMid:19790218 [ Links ]
44. Weber D, Tarselli MA, Gagné MR. Mechanistic surprises in the gold(I)-catalyzed intramolecular hydroarylation of allenes. Angew Chem Int Ed. 2009;48:5733-5736. doi:10.1002/anie.200902049, PMid:19562821, PMCid:2978329 [ Links ]
45. Cheong PH, Morganelli P, Luzung MR, Houk KN, Toste FD. Gold-catalyzed cycloisomerization of 1,5-allenynes via dual activation of an ene reaction. J Am Chem Soc. 2008;130:4517-4526. doi:10.1021/ja711058f, PMid:18327944, PMCid:2995695 [ Links ]
46. Hashmi ASK, Schuster A, Rominger F. Gold catalysis: Isolation of vinylgold complexes derived from alkynes. Angew Chem Int Ed. 2009;48:8247-8249. doi:10.1002/anie.200903134, PMid:19768822 [ Links ]
47. Hashmi ASK, Ramamurthi TD, Rominger F. On the trapping of vinylgold intermediates. Adv Synth Catal. 2010;352:971-975. doi:10.1002/adsc.201000011 [ Links ]
48. Strasser CE, Stander-Grobler E, Schuster O, Cronje S, Raubenheimer HG. Preparation of remote NHC complexes of rhodium(I) and gold(I) by ligand transfer. Eur J Inorg Chem. 2009;1905-1912. doi:10.1002/ejic.200801180 [ Links ]
49. Raubenheimer H, Cronje S. Carbene complexes of gold: Preparation, medical application and bonding. Chem Soc Rev. 2008;37:1998-2011. doi:10.1039/b708636a, PMid:18762843 [ Links ]
50. Solorio-Alvarado CR, Echavarren AM. Gold-catalyzed annulation/fragmentation: Formation of free gold carbenes by retro-cyclopropanation. J Am Chem Soc. 2010;132:11881-11883. doi:10.1021/ja104743k, PMid:20698529 [ Links ]
51. Binger P, Müller P, Benn R, et al. Vinylcarbene complexes of titanocene. Angew Chem Int Ed Engl. 1989;28:610-611. doi:10.1002/anie.198906101 [ Links ]
52. Nguyen ST, Johnson LK, Grubbs RH. Ring-opening metathesis polymerization (ROMP) of norbornene by a group VIII carbene complex in protic media. J Am Chem Soc. 1992;114:3974-3975. doi:10.1021/ja00036a053 [ Links ]
53. Seidel G, Mynott R, Fürstner A. Elementary steps of gold catalysis: NMR spectroscopy reveals the highly cationic character of a "gold carbenoid". Angew Chem Int Ed. 2009;48:2510-2513. doi:10.1002/anie.200806059, PMid:19248070 [ Links ]
54. Schuster O, Yang L, Raubenheimer HG, Albrecht M. Beyond conventional N-heterocyclic carbenes: Abnormal, remote, and other classes of NHC ligands with reduced heteroatom stabilization. Chem Rev. 2009;109:3445-3478. doi:10.1021/cr8005087, PMid:19331408 [ Links ]
55. Raubenheimer HG, Esterhuysen MW, Timoshkin A, Chen Y, Frenking G. Electrophilic addition of Ph3PAu+ to anionic alkoxy Fischer-type carbene complexes: A novel approach to metal stabilised bimetallic vinyl ether complexes. Organometallics. 2002;21:3173-3181. doi:10.1021/om020048g [ Links ]
56. Raubenheimer HG, Esterhuysen MW, Frenking G, et al. Aurolysis of a-C-deprotonated group 6 aminocarbene and thiocarbene complexes with Ph3PAu+. Dalton Trans. 2006; 4580-4589. doi:10.1039/b607613k, PMid:17016569
57. Fürstner A, Morency L. On the nature of the reactive intermediates in gold-catalyzed cycloisomerization reactions. Angew Chem Int Ed. 2008;47:5030-5033. doi:10.1002/anie.200800934, PMid:18504793 [ Links ]
58. Hashmi SAK. "High noon" in gold catalysis: Carbene versus carbocation intermediates. Angew Chem Int Ed. 2008;47:6754-6756. doi:10.1002/anie.200802517, PMid:18683174 [ Links ]
59. Roth KE, Blum S. Relative kinetic basicities of organogold compounds. Organometallics. 2010;29:1712-1716. doi:10.1021/om901101f [ Links ]
60. Smith MB, March J. March's advanced organic chemistry: Reactions, mechanisms and structure. 5th ed. New York: John Wiley & Sons, 2001; p. 329. [ Links ]
 Correspondence to:
Correspondence to:
Helgard Raubenheimer
Postal address:
Private Bag X1
Matieland 7602, South Africa
email: hgr@sun.ac.za
Received: 05 Oct. 2010
Accepted: 06 Dec. 2010
Published: 16 Mar. 2011
© 2011. The Authors. Licensee: OpenJournals Publishing. This work is licensed under the Creative Commons Attribution License.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}