










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Science
versión On-line ISSN 1996-7489
versión impresa ISSN 0038-2353
S. Afr. j. sci. vol.106 no.5-6 Pretoria may./jun. 2010
RESEARCH ARTICLE
An assessment of the atmospheric nitrogen budget on the South African Highveld
Kirsten S. CollettI; Stuart J. PikethI; Kristy E. RossII
IClimatology Research Group, University of the Witwatersrand, Johannesburg, South Africa
IIEskom Generation, Megawatt Park, Johannesburg, South Africa
ABSTRACT
Atmospheric reactive nitrogen concentrations on the South African Highveld have become a growing concern, with satellite images indicating very high nitrogen dioxide (NO2) concentrations in the region. This study investigated the nitrogen budget on the Highveld through the analysis of the concentration of the atmospheric nitrogen species on a temporal scale as well as the atmospheric conversion, transport and removal of these species. Data were collected at Eskom's Elandsfontein ambient air quality monitoring site, which is centrally located on the industrialised Highveld. A year's dataset from 2005 and 2006 was analysed and it was found that nitrogen oxide (NOx) concentrations were higher in winter as a result of stable atmospheric conditions, as well as prevalent westerly and north-westerly airflow, which transported emissions directly from the nearby power station sources to the monitoring site. Nitrate (NO3) concentrations also peaked during winter, with a distinct biomass burning peak during August 2005. Diurnally, NOx concentrations indicated a tall-stack industrial source, where concentrations peaked at midday. The NO3 concentrations were higher at night than during the day; during the day the NO3 radical is rapidly photolysed and nitrates cannot be produced. Case studies indicated that the conversion rate of nitric oxide (NO) to NO2 was highly variable as a result of varying atmospheric factors, which include time of day, dispersion, stability and regional atmospheric chemistry. These rates ranged from 11% to 59% per hour. Rates of dry deposition of NO, NO2 and NO3 were generally higher during winter as a result of higher atmospheric concentrations and increased atmospheric stability. Nitrogen was predominantly deposited as NO2 throughout the year, except during spring when NO3 deposition dominated. The total annual amount of nitrogen that was deposited to the Mpumalanga Highveld region was in the range of 6.7 kg/ha - 13.1 kg/ha per year, which is well below the stipulated critical load value. Such deposition, therefore, should not pose significant threats to the natural environment on the Highveld. Between 4% and 15% of the total emitted nitrogen from power generation on the Highveld was deposited to the surface via wet and dry deposition. The remainder was advected out of the region.
Keywords: atmospheric conversion; deposition; diurnal variations; nitrate; nitrogen oxides; seasonal variations
INTRODUCTION
Molecular nitrogen is highly abundant in the atmosphere and exists in a natural balance. Anthropogenic activities, however, have disrupted the natural nitrogen cycle via the production of reactive and sometimes toxic nitrogen-containing compounds. At elevated concentrations, such compounds may lead to severe health and environmental effects through their transport and deposition to the earth's surface.1,2
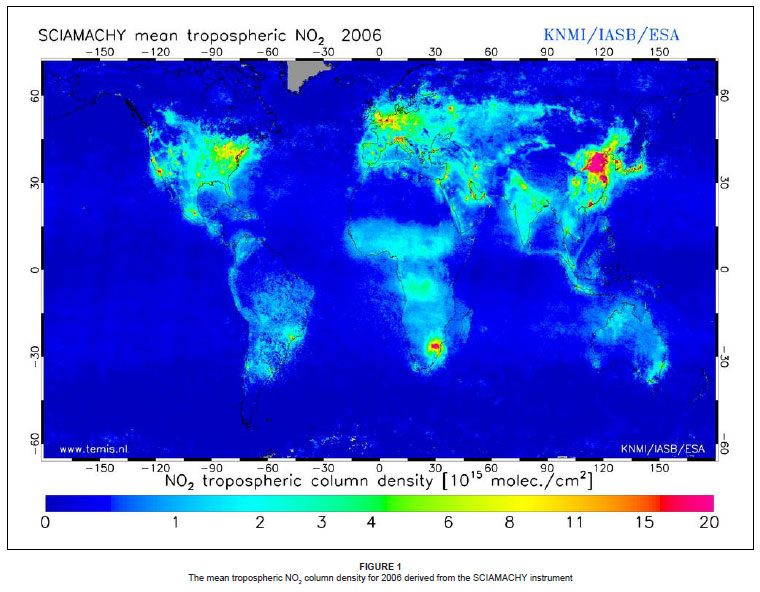
The South African Highveld is a highly industrialised area containing rich coal reserves. It is home to nine coal-fired power stations, which are in close proximity to one another, resulting in very high emission densities in the region. The area is also home to heavy industry, located within small towns, as well as the petrochemical industries in Sasolburg and Secunda. These industrial activities are all responsible for disturbing the nitrogen budget in the region.3,4,5,6 According to satellite retrievals, such as the Global Ozone Monitoring Experiment (GOME) and the Scanning Imaging Absorption Spectrometer for Atmospheric Chartography (SCIAMACHY), the South African Highveld is an area of highly elevated nitrogen dioxide (NO2) concentrations (Figure 1). Although satellite-based instruments are effective for recording trace gas concentrations with fixed spatial and temporal resolutions over large timescales, the area of elevated NO2 concentration over the Highveld region has been identified as one of the outliers in the satellite dataset.7 This implies uncertainty, particularly where it has been found that field research does not correlate with the satellite data, and so further investigation is required.
A relatively comprehensive understanding of the sulphur budget on the Mpumalanga Highveld has been acquired.8 Various sulphur deposition studies have also been performed by Zunckel et al. over the years.9,10,11 However, there is limited knowledge regarding atmospheric nitrogen on the Highveld, necessitating further research in the field. This study provides insight into the atmospheric nitrogen budget on the South African Highveld by analysing the atmospheric concentration, transport and deposition of various nitrogen species in the region.
METHODS
Study area
The industrialised Highveld plateau region in north-eastern South Africa extends across parts of Gauteng, the Free State and Mpumalanga provinces at about 1400 m - 1700 m above sea level.3,4,12 About 70% of the Highveld area is covered by grassland and the rest is utilised for agricultural, urban and industrial activities. Being a highly industrialised area (accounting for about 75% of South Africa's industrial activity), this region accounts for about 90% of the nitrogen oxide (NOx) emitted in South Africa.4,12
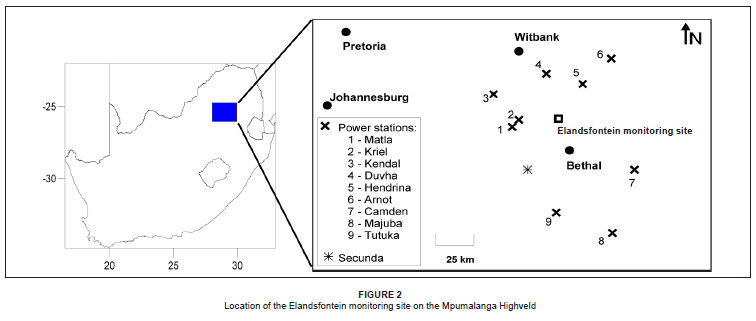
Field measurements were conducted at Eskom's Elandsfontein ambient air quality monitoring site in central Mpumalanga (Figure 2), which is ideally situated in the hub of the industrialised region, allowing for an intensive study of industrial nitrogen emissions.
Data and instrumentation
The Elandsfontein monitoring station houses various trace gas, particulate and meteorological instruments. Concentrations of NO2, nitric oxide (NO), nitrate (NO3), ammonia (NH3), ozone (O3), sulphur dioxide (SO2), hydrogen sulphide (H2S) and black carbon were continuously monitored and meteorological information was recorded at the site. Data from a one-year period, from 1 April 2005 to 31 March 2006, were utilised in the analysis. Owing to instrumentation constraints, only one year of data has been analysed in this study. This dataset, however, provides statistically significant seasonal patterns of nitrogen compounds because it is the only dataset that can examine NO3 formation in the region.
A TSI Model 17C Chemiluminescence NH3 analyser (Thermo Environmental Instruments Inc., Franklin, USA) was utilised to measure NOx and NH3 concentrations whilst particulate nitrates were measured using a Rupprecht and Patashnick Series 8400N Ambient Particulate Nitrate Monitor (Thermo Electron Corporation, New York, USA). Ozone concentrations were recorded using a TSI UV Photometric O3 analyser (Thermo Environmental Instruments Inc.) and meteorological parameters were measured with various meteorological instruments (mainly RM Young, Traverse City, USA) that were mounted on a microwave mast located at the site. During the one-year study period, the gas analysers and particulate nitrate monitor were calibrated on a monthly basis. The instrumentation calibration was intended to eliminate or reduce bias in the instruments' readings. During the calibration, reference standards with known values for selected points covering the range of interest were measured with the instrument in question. A functional relationship was then established between the values of the standards and the corresponding measurements in order to correct for instrument bias. Range checks were also performed, where any values that were not within the measured operating range of the instrument were removed. After power cuts, several hours were allowed for instrumentation warm-up and coinciding data were eliminated. At Elandsfontein, none of the instruments showed any zero drift. Any span shifts were corrected during calibration and data were subsequently corrected for the previous month based on the zero and span checks.
Methodology
Rates of conversion of NO to NO2 were calculated using various case-study days when the wind was blowing directly from an industrial source towards Elandsfontein, as well as when high NO2 concentrations were experienced. A geometric sequence was utilised to calculate the percentage of NO that converts per hour as an air mass travels downwind from its source. The literature assumes that the initial NO to NO2 ratio on release into the atmosphere is about 95:5.13 According to Eskom, however, the ratio of NO to NO2 released from their power station stacks is 98:2 (Ross KE 2008, personal communication, June 30); therefore, this latter ratio was used in the calculations. The final concentration was calculated as a percentage that was based on the ratio of NO to NO2 recorded at the monitoring site.
Conversion rates for NO to NO2 were calculated during the day only, because without the presence of sunlight, O3 is unable to be produced and so one of the main conversion pathways of NO2 stops. Also, any industrial NOx that is produced at night is trapped above the natural inversion layer. This NOx can only be detected at the monitoring site the next day, after the inversion has dissipated. Vertical mixing within the inversion layer is poor, but at times a strong horizontal wind shear is present. At night, particularly in winter, NOx may also be advected away in a low-level jet. These jets develop above the natural inversion layer, at about 150 m - 300 m above the ground, and have been found to transport air masses and pollutants distances of up to 400 km in one night.3,14,15 Such phenomena provide a highly variable indication of the rate of conversion due to the different residence times in the atmosphere.
Conversion rate calculations are very challenging because they are based on numerous assumptions. These assumptions include, (1) that all the NO and NO2 comes from one specific source and (2) that the rate of conversion was uniform for each hour that the air mass transports downwind. Other factors that need to be considered when calculating conversion rates are: wind speed, dispersion in the region, stability, transport from other areas, regional atmospheric chemistry and time of day.
Dry deposition processes are very significant on the Highveld plateau, as the area receives mostly summer rainfall on average only 60 days per year.9 Dry deposition has therefore been the focus of this research. Wet deposition rates are, however, also included from a previous study performed by Mphepya16 in 2002 over the same area.
The inferential method was employed to calculate rates of dry deposition. The inferential method calculates the dry deposition flux (F) using the atmospheric concentration (C) and the deposition velocity ( vd ) of a certain species through the equation F = -vdC.
Active nitrogen concentration measurements at Elandsfontein were utilised in the calculations along with vd values taken from the literature.2,17,18,19,20,21,22,23,24,25 These vd values range from 0 cm/s to 1.5 cm/s for NO and NO2 and 0.12 cm/s to 1.2 cm/s for NO3. To compensate for the large variations, the mean, maximum and minimum of these vd values were utilised in the calculations. This provides a more representative measure of how the possible flux values are spread. Thus vd is described as a parameterisation of the rate of transfer of a pollutant from the atmosphere to the receptor surface,26,27,28,29,30,31 where vd is expressed as the reciprocal of a series of the aerodynamic, boundary layer and canopy resistances. Both vd and C are height-dependent and the negative sign indicates a downward flux.2,30,32,33,34,35
The inferential model has been found to compare well with other micrometeorological techniques10,25,26,27 and has been utilised during similar deposition studies on the Highveld.9,10,11,16,36 Most recently, Lowman36 utilised the inferential technique to analyse nitrogen deposition to a grassland area and a forested area on the Mpumalanga Highveld.
RESULTS AND DISCUSSION
Seasonal variations
For the study period, NO concentrations peaked in winter (May and June) (Figure 3) with no other significant trends. The prevalent westerly and north-westerly airflow at this time, associated with westerly waves and anticyclonic circulations,37 transported NO directly from the nearby power station sources to the monitoring site. This winter peak may also be a result of stable conditions, which prevented upward mixing.
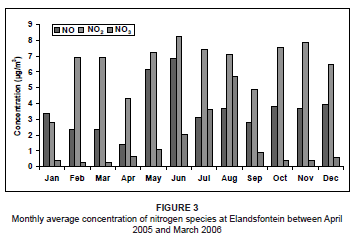
The NO2 concentrations during this period were variable from month to month, with lower concentrations during January, April and September. The highest concentrations occurred during winter, as a result of very stable atmospheric conditions that limited the amount of atmospheric dilution. The NO3 concentrations were relatively low throughout the year, but showed a marked peak during July and August. This peak could have resulted from atmospheric accumulation as a result of stable conditions and no rainfall. This was very similar to what was found by Scheifinger and Held during a study on the Highveld in 1997.3 The winter peak may also have been augmented by biomass burning emissions in the region at the time. Biomass fires produce high concentrations of NOx as a result of the high nitrogen content of the fuel.38 The NO2 concentrations during this time were also expected to be at a maximum as a result of this biomass burning. However, NO2 readily oxidises due to the high occurrence of O3 in the atmosphere (Figure 4) to produce higher NO3 concentrations. As a result of these high O3 levels during July and August, it was likely that NO rapidly oxidised into NO2, resulting in much lower NO concentrations than during May and June, provided that all other atmospheric conditions remained the same.
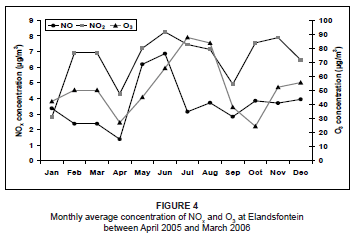
For the study period, O3 concentrations peaked during winter (Figure 4). This was a result of elevated NOx concentrations that were rapidly photolysed in the presence of sunlight to produce O3. For the context of this paper, this is a highly simplified explanation of O3 formation, as other species are also involved in the chemistry. NOx acts as the catalyst for O3 production when hydrocarbons, volatile organic compounds (VOCs) and carbon monoxide (CO) are oxidised.2,39,40,41 High O3 concentrations during August may be attributed to biomass burning in the region. Previous studies have discovered peaks in O3 concentrations during late winter and spring as a result of biomass fires, which produce high concentrations of O3 precursors.6,42,43
Diurnal variations
Using the hourly averages of all the days for the study period, the diurnal pattern of NO and NO2 indicated low concentrations during the night and a peak around midday (Figure 5). This was a result of surface inversions which developed at night. Power generation and petrochemical industrial stacks are 200 m - 300 m in height and emit pollutants well above this natural inversion.3 Pollutants are unable to move to the surface at night. After sunrise, convective mixing is initiated and the surface inversion breaks down, allowing pollutants to be transported to ground level. The O3 concentrations were low at night and peaked during the day, when both NO2 and sunlight were available. The diurnal trends in concentration, however, do not reflect the expected NOx-O3 photochemical behaviour. During the O3 peak, a major decrease in NO2 concentration was not experienced, so it could be assumed that some of the O3 was transported into the area or that NO2 was continually forming. There was also a disproportionate amount of O3 produced in the atmosphere in relation to the amount of NO2 that was available. This is probably due to the fact that not all of the O3 was a direct result of NO2 dissociation; the O3 exists at background levels of about 40 µg/m3. It also needs to be considered, however, that concentrations of other O3 precursors (like VOCs, CO and hydrocarbons) were not measured and analysed during this study and may also have an impact on the unusual O3 concentration measurements.

To aid in confirming that the NOx emissions recorded at Elandsfontein were predominantly a result of industrial sources, black carbon and SO2 concentrations were also plotted in Figure 5. The SO2 concentrations peaked at midday, after the nocturnal surface inversion had dissipated and pollutants could be transported to the surface, clearly indicating the industrial origin of the SO2 emissions. No peaks in SO2 and black carbon concentrations occurred during the morning or early evening, confirming that the emissions monitored at Elandsfontein did not originate from domestic coal fires in the region.
The NO3 concentrations were higher at night than they were during the day (Figure 6), because the NO3 radical is rapidly photolysed during the day and this prohibits the formation of nitrates.2,44,45

Atmospheric conversion
Rates of conversion of NO to NO2 ranged from 11% to 59% per hour. It was observed that the higher the concentration of O3, the higher the NO to NO2 conversion rate; the more O3 that is available for oxidation, the greater the capacity for NO2 formation. Ambient temperature was found to not have had a direct effect on the rate of conversion. Although the range of calculated conversion rates was large, these values corresponded well with the maximum NO to NO2 conversion rate of 30% per hour, as calculated by Hewitt.13 Gertler et al.46, however, found much lower conversion rates of about 8% per hour.
Conversion rates of NO to NO3 were not as easily calculated because NO3 only forms at night. A peak in NO3 concentration at night is not related to an industrial plume, as these plumes are trapped above the natural inversion layer at night and were thus not detected at the monitoring site. Any peaks during the night could be attributed to aged industrial plumes or plumes from non-industrial NOx sources that are close to the surface.
Case study 1: Elevated NOx concentrations associated with coal-fired power station sources
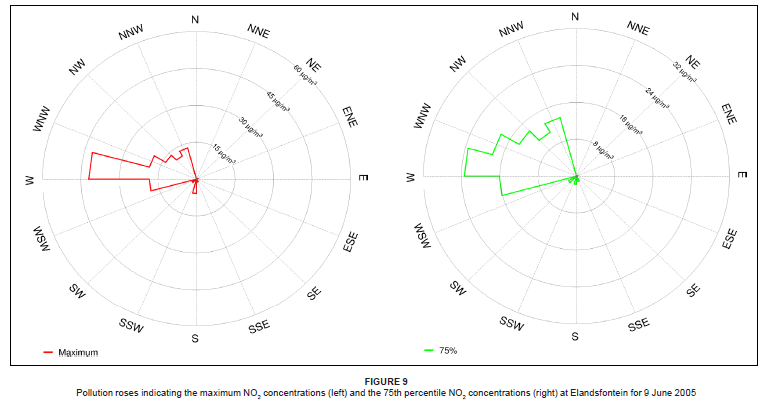
On 9 June 2005, the diurnal concentrations of NOx and SO2 clearly indicated a tall-stack industrial source, with concentrations peaking around midday and diminishing at night (Figure 7). A sharp drop in O3 concentration occurred at midday, which is highly unusual. This decrease strongly reflected the occurrence of a direct plume impact at the site. Background O3 was used up in the reaction to produce NO2. A second minor industrial peak in NOx and SO2 was experienced between 15:00 and 17:00. This peak was not of vehicle or domestic fuel burning origin, since the black carbon concentration was low and the dominant airflow was from Kriel and Matla at this time. A slight morning peak in SO2, NO2 and black carbon (between 08:00 and 10:00) suggests the influence of domestic coal burning. Airflow was from the west-south-west at this time, transporting emissions from Thubelihle township that is situated just outside the town of Kriel, 12 km from Elandsfontein.

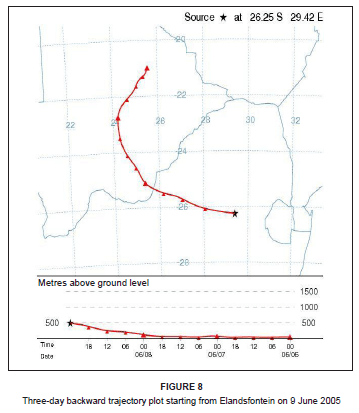
On this day, westerly airflow dominated in association with the anticyclonic circulation of an upper level high pressure system. Both the trajectory analysis (Figure 8) and pollution roses (Figure 9) suggest that the high NOx concentrations could be attributed to the Kriel, Matla and Kendal power stations that lie to the west of Elandsfontein. The average rate of conversion of NO to NO2 in this power station plume was calculated to be 34% per hour (wind speed, 6 m/s; distance from source, 25 km).
Case study 2: Elevated NOx concentrations associated with a petrochemical source
On 11 April 2005, higher NOx and SO2 concentrations during the day once again indicated a tall-stack source (Figure 10). The O3 concentrations remained high during the day as a result of available precursors and sunlight. During the NOx peak, O3 concentrations decreased to 0 µg/m3 as O3 was used up in the production of NO2 (O3 is not emitted directly into the atmosphere from the source). From 15:00, O3 concentrations increased until a maximum was reached at 20:00. Since all the other pollutants did not increase accordingly at this time, it was assumed that this O3 was not a result of local sources but may have been a result of long-range transport from other regions.

At the time of the plume impact at Elandsfontein the ratio of NO to NO2 was approximately 1, which indicated that the plume had aged somewhat. On this day, the average conversion rate of NO to NO2 was between 21% and 30% per hour (wind speed, 0.9 m/s - 2.2 m/s; distance from source, 40 km).
The pollution roses for 11 April 2005 indicated that the highest NO2 concentrations were associated with flow from the Kriel, Matla and Kendal power stations (Figure 11). The pattern in the SO2 concentrations, however, did not reflect a typical power station plume impact. The concentrations started to increase at 10:00 and reached a maximum between 13:00 and 17:00. The elevated concentrations (between 50 µg/m3 and 100 µg/m3) remained over the station until 20:00. The H2S concentrations spiked at the same time as the other pollutants; H2S is a primary emission from the Secunda plant.47 The trajectory analysis for 11 April 2005 confirmed that the high concentrations observed during the day may have originated from Secunda in the south-west (Figure 12). This trajectory depicts upper-level airflow (at 500 m above ground level), whereas the pollution roses indicate surface airflow. The higher concentrations may then be associated with the period of upper-level anticyclonic stability, resulting in south-westerly transport of industrial emissions from Secunda towards Elandsfontein. This period of anticyclonic stability over the Highveld was also conducive to the accumulation of pollution between 10:00 and 20:00.

Deposition
Seasonal deposition patterns
As a result of the large range in vd values in the literature, the expected uncertainties in the dry deposition flux estimates when utilising the inferential model are between 30% and 50%.48 Calculated dry deposition flux rates for the three nitrogen species were very different when utilising the mean vd values from the literature in the inferential calculations (Figure 13). The NO dry deposition rates were much higher in winter months (peaking in June) than in summer months. Dry deposition rates of NO2 were highly variable throughout the year, with a general peak during winter. Particularly high rates of dry deposition of NO2 occurred during March. The rates of NO3 deposition peaked quite considerably during late winter, as a result of accumulation and fallout to the surface. The peak NO3 deposition rate in August was considerably higher than the NOx deposition rates. Predominant winter deposition of all three species may be a result of increased atmospheric stability, preventing transport out of the region, in winter.

Throughout the year, nitrogen was predominantly deposited in the form of NO2, except during spring, when deposition in the form of NO3 dominated. Similarly, Hesterberg et al.22 found higher rates of NOx and NO3 deposition during winter as a result of higher concentrations at this time. According to Meyers et al.26, rates of NO3 deposition to various grassland and forested sites in the USA were also higher during spring.
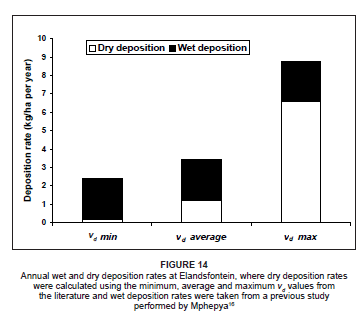
To evaluate the uncertainty in the depositional flux values as a result of the variable vd values from the literature, the depositional fluxes were calculated using the minimum, average and maximum vd values for each of the species. A similar approach was adopted by Lowman36 during a nitrogen deposition study on the Highveld. Figure 14 indicates the range of possible dry depositional flux values for the Highveld, as well as the annual wet deposition flux value obtained from Mphepya's16 study. On the Highveld, rates of dry deposition were calculated to be considerably lower than the wet deposition rates when using the minimum and average vd values in the calculations. When using the maximum vd values in the deposition calculations, dry deposition exceeded wet deposition in the region considerably.
Total deposition to the Highveld
The annual rate of dry deposition of nitrogen to the Highveld from April 2005 to March 2006 ranged from 0.18 kg N/ha per year to 6.6 kg N/ha per year (Figure 14), depending on which vd value was utilised. When combined with the wet deposition rates, the total nitrogen deposited to the region was in the range of 2.4 kg N/ha per year to 8.8 kg N/ ha per year. This rate was well below the rates calculated in previous studies in various grassland areas around the world.22,36,48,49,50 In this study, however, only NO, NO2 and NO3 were considered in the calculations. Even though NH3 is also a very important compound in nitrogen deposition,22,36,51 there was, unfortunately, no NH3 data from Elandsfontein available during the study period, thus NH3 could not be included in the deposition calculations. By incorporating the NH3 deposition rate value of 4.3 kg N/ha per year from Lowman's study conducted in the same area (as a baseline for NH3 deposition on the Highveld), the resultant range in dry deposition values at Elandsfontein was between 4.5 kg N/ha per year and 10.9 kg N/ha per year. If the wet deposition rates were also included, the total nitrogen deposition was between 6.7 kg N/ha per year and 13.1 kg N/ha per year. These values are much closer to the ranges calculated during previous nitrogen studies, providing a more accurate interpretation of total nitrogen deposition to the region.
Critical loads provide a measure of how elevated concentrations of a pollutant will potentially negatively affect sensitive elements of an environment.52 This value is determined based on the vegetation type in the region and the soil characteristics. The critical load for nitrogen for grasslands has been set at 15 kg N/ha per year.53 For the Highveld region, for the study period, the values of 6.7 kg N/ha per year and 13.1 kg N/ha per year were well below the stipulated critical load and hence such deposition should not pose significant threats to the natural environment. Josipovic et al.54 also discovered similar results utilising passive samplers, where levels of NO2 deposition across the Highveld were well below the critical level.
Using emissions data from Eskom's Annual Reports for 2005 and 2006 along with the inferential model calculations, it was determined that between 4% and 15% of the total emitted industrial nitrogen from power generation on the Highveld was deposited to the surface via wet and dry deposition processes. It was assumed that the remaining nitrogen (between 85% and 96%) was advected out of the region. These values, however, may underestimate the total amount of nitrogen deposited to the region, as nitrogen emission sources from other industries, vehicles and domestic combustion, as well as other aerosol forms of nitrogen, were not included in the calculations. These emissions may potentially increase the amount of nitrogen that is deposited to the surface.
Although the amount of nitrogen deposited to the surface was well below the critical load, the percentage of nitrogen that remained in the atmosphere and was advected out of the region is significant. This increase in atmospheric nitrogen causes a positive balance in the atmospheric nitrogen budget. Such an increase results in questions about the impact of this added nitrogen on the troposphere and may begin to confirm the nitrogen 'hotspot' detected by GOME and SCIAMACHY.
CONCLUSION
Anthropogenic activities have disturbed the natural nitrogen cycle on the South African Highveld. Increased atmospheric concentrations of NO, NO2 and NO3 have led to a positive nitrogen balance as more of these molecules remain in the atmosphere than are deposited to the surface. For the observed year, NO, NO2 and NO3 concentrations peaked during winter. The diurnal signals of NO and NO2 clearly indicated tall-stack industrial sources, where concentrations peaked at midday and diminished at night. NO3 concentrations were higher at night and lower during the day, because, in the presence of sunlight, the NO3 radical rapidly photolyses and nitrates cannot be produced.
Rates of conversion of NO to NO2 were calculated for case-study days when the wind was blowing directly from an industrial source towards the monitoring station. The estimated rates of conversion ranged from 11% to 59% per hour although, given the uncertainties and assumptions in the methodology used, the actual range could be higher than this. Using the inferential method, rates of dry deposition were calculated to be higher during the winter months. Deposition was predominantly in the form of NO2 throughout the year, except for spring when NO3 deposition dominated.
The total amount of nitrogen deposited to the Mpumalanga Highveld region was in the range of 6.7 kg N/ha per year to 13.1 kg N/ha per year, which is below the stipulated critical load value for grasslands of 15 kg N/ha per year. For the whole industrial Highveld area, it was found that less than 15% of the total emitted nitrogen from power generation was deposited to the surface via wet and dry deposition. The rest remained in the atmosphere and was advected out of the region.
ACKNOWLEDGEMENTS
The collection of the air quality data at the Elandsfontein monitoring station was funded by Eskom's Sustainability and Innovation Department, Corporate Services Division. Eric Lynch, Neil Snow and the air quality monitoring team at Eskom are thanked for providing the data. KNMI/IASB/ESA are also acknowledged for the use of their SCIAMACHY satellite imagery, obtainable from http://www.temis.nl
REFERENCES
1. Galloway JN, Levy II H, Kasibhatla PS. Year 2020: Consequences of population growth and development on deposition of oxidized nitrogen. Ambio. 1994;23(2):120-123. [ Links ]
2. Seinfeld JH, Pandis SN. Atmospheric chemistry and physics: From air pollution to climate change. New York: Wiley; 1998. [ Links ]
3. Scheifinger H, Held G. Aerosol behaviour on the South African Highveld. Atmos Environ. 1997;31(21):3497-3509. [ Links ]
4. Freiman MT, Piketh SJ. Air transport into and out of the industrial Highveld region of South Africa. J Appl Meteorol. 2003;42:994-1002. [ Links ]
5. Held G, Snyman GM, Scheifinger H. Seasonal variations and trends of atmospheric particulates on the South African Highveld. Clean Air J. 1993;8(8):4-11. [ Links ]
6. Held G, Gore BJ, Surridge AD, Tosen GR, Turner CR, Walmsley RD, editors. Air pollution and its impacts on the South African Highveld. Cleveland: Environmental Scientific Association; 1996. [ Links ]
7. Toenges-Schuller N, Stein O, Rohrer F, et al. Global distribution pattern of anthropogenic nitrogen oxide emissions: Correlation analysis of satellite measurements and model calculations. J Geophys Res. 2006;111(D05312), doi:10.1029/2005JD006068. [ Links ]
8. Igbafe AI. Resolving the atmospheric sulphur budget over the Elandsfontein area of the Mpumalanga Highveld. PhD thesis, University of the Witwatersrand, Johannesburg; 2007. [ Links ]
9. Zunckel M, Turner CR, Wells RB. Dry Deposition of sulphur on the Mpumalanga Highveld: A pilot study using the inferential method. S Afr J Sci. 1996;92:485-491. [ Links ]
10. Zunckel M, Piketh S, Freiman T. Dry deposition of sulphur at a high-altitude background station in South Africa. Water Air Soil Pollut. 1999;115:445-463. [ Links ]
11. Zunckel M, Robertson L, Tyson PD, Rodhe H. Modelled transport and deposition of sulphur over southern Africa. Atmos Environ. 2000;34:2797-2808. [ Links ]
12. Wenig M, Spichtinger N, Stohl A, et al. Intercontinental transport of nitrogen oxide pollution plumes. Atmos Chem Phys. 2003;3:387-393. [ Links ]
13. Hewitt CN. The atmospheric chemistry of sulphur and nitrogen in power station plumes. Atmos Environ. 2001;35:1155-1170. [ Links ]
14. Zunckel M, Held G, Preston-Whyte RA, Joubert A. Low-level wind maxima and transport of pyrogenic products over southern Africa. J Geophys Res. 1996;101(D19):23745-23755. [ Links ]
15. Smith TB, Blumenthal DL, Anderson JA, Vanderpol AH. Transport of SO2 in power plant plumes: Day and night. Atmos Environ. 1978;12:605-611. [ Links ]
16. Mphepya JN. Atmospheric deposition characteristics of sulphur and nitrogen compounds in South Africa. PhD thesis, Potchefstroomse Universiteit vir Christelike Hoer Onderwys, Potchefstroom; 2002. [ Links ]
17. Wesely ML, Eastman JA, Stedman DH, Yalvac ED. An eddy correlation measurement of NO2 flux to vegetation and comparison to O3 flux. Atmos Environ. 1982;16(4):815-820. [ Links ]
18. Duyzer JH, Meyer GM, Van Aalst RM. Measurement of dry deposition velocities of NO, NO2 and O3 and the influence of chemical reactions. Atmos Environ. 1983;17(10):2117-2120. [ Links ]
19. Heubert BJ, Luke WT, Delany AC, Brost RA. Measurements of concentrations and dry surface fluxes of atmospheric nitrates in the presence of ammonia. J Geophys Res. 1988;93(D6):7127-7136. [ Links ]
20. Hanson PJ, Lindberg SE. Dry deposition of reactive nitrogen compounds: A review of leaf, canopy and non-foliar measurements. Atmos Environ. 1991;25A(8):1615-1634. [ Links ]
21. Coe H, Gallagher MW. Measurements of dry deposition of NO, to a Dutch heathland using the eddy-correlation technique. Q J R Meteorol Soc. 1992;118:767-786. [ Links ]
22. Hesterberg R, Blatter A, Fahrni M, et al. Deposition of nitrogen containing compounds to an extensively managed grassland in central Switzerland. Environ Pollut. 1996;91(1):21-34. [ Links ]
23. Horvath L, Nagy Z, Weidinger T. Estimation of dry deposition velocities of nitric oxide, sulphur dioxide and ozone by gradient method above short vegetation during the Tract campaign. Atmos Environ. 1998:32(7):1317-1322. [ Links ]
24. Zapletal M. Atmospheric deposition of nitrogen compounds in the Czech Republic. Environ Pollut. 1998;102(S1):305-311. [ Links ]
25. Watt SA, Wagner-Riddle C, Edwards G, Vet RJ. Evaluating a flux-gradient approach for flux and deposition velocity of nitrogen dioxide over short-grass surfaces. Atmos Environ. 2004;38:2619-2626. [ Links ]
26. Meyers TP, Hicks BB, Hosker RP, Womack JD, Satterfield LC. Dry deposition inferential measurement techniques - II. Seasonal and annual deposition rates of sulfur and nitrate. Atmos Environ. 1991;25A(10):2361-2370. [ Links ]
27. Brook JR, Di-Giovanni F, Cakmak S, Meyers TP. Estimation of dry deposition velocity using inferential models and site specific meteorology - Uncertainty due to siting of meteorological towers. Atmos Environ. 1997;31(23):3911-3919. [ Links ]
28. Clarke JF, Edgarton ES, Martin BE. Dry deposition calculations for the Clean Air Status and Trends Network. Atmos Environ. 1997;31(21):3667-3678. [ Links ]
29. Asman WAH, Sutton MA, Schjorring JK. Ammonia: Emission, atmospheric transport and deposition. New Phytol. 1998;139:27-48. [ Links ]
30. Wesley ML, Hicks BB. A review on the current status of knowledge on dry deposition. Atmos Environ. 2000;34:2261-2282. [ Links ]
31. Schmidt M, Thoni L, Waldner P, Thimonier A. Total deposition of nitrogen on Swiss long-term forest ecosystem research (LWF) plots: Comparison of the throughfall and the inferential method. Atmos Environ. 2005;39:1079-1091. [ Links ]
32. Baumgardner RE, Lavery TF, Rogers CM, Isil SS. Estimates of the atmospheric deposition of sulfur and nitrogen species: Clean air status and trends network, 1990-2000. Environ Sci Technol. 2002;36:2614-2629. [ Links ]
33. Gao Y. Atmospheric nitrogen deposition in Barnegat Bay. Atmos Environ. 2002;36:5783-5794. [ Links ]
34. Marner BB, Harrison RM. A spatially refined monitoring based study of atmospheric nitrogen deposition. Atmos Environ. 2004;38:5045-5056. [ Links ]
35. Yang H, Hsieh L, Cheng S. Determination of atmospheric nitrate particulate size distribution and dry deposition velocity for three distinct areas. Chemosphere. 2005;60:1447-1453. [ Links ]
36. Lowman GRP. Deposition of nitrogen to grassland versus forested areas in the vicinity of Sabie, Mpumalanga, South Africa. MSc dissertation, University of the Witwatersrand, Johannesburg; 2003. [ Links ]
37. Tyson PD, Garstang M, Swap R, Kållberg P, Edwards M. An air transport climatology for subtropical southern Africa. Int J Climatol. 1996;16:265-291. [ Links ]
38. Warneck P. Chemistry of the natural atmosphere, International Geophysics Series vol. 41. San Diego: Academic Press Inc.; 1988. [ Links ]
39. Jacob DJ, Heikes BG, Fan SM, et al. Origin of ozone and NOx in the tropical troposphere: A photochemical analysis of aircraft observations over the South Atlantic basin. J Geophys Res. 1996;101(D19):24235-24250. [ Links ]
40. Ryerson TB, Buhr MP, Frost GJ, et al. Emissions lifetimes and ozone formation in power plant plumes. J Geophys Res. 1998;103(D17):22569-22583. [ Links ]
41. Colls J. Air pollution. Taylor and Francis: New York; 2002. [ Links ]
42. Hobbs PV, Sinha P, Yokelson RJ, et al. Evolution of gases and particles from a savanna fire in South Africa. J Geophys Res. 2003;108(D13):8485. [ Links ]
43. Zunckel M, Venjonoka K, Pienaar JJ, et al. Surface ozone over southern Africa: Synthesis of monitoring results during the Cross Border Air Pollution Impact Assessment project. Atmos Environ. 2004;38:6139-6147. [ Links ]
44. Wayne RP, Barnes I, Biggs P, et al. The nitrate radical: Chemistry, physics and the atmosphere. Atmos Environ. 1991;25A(1):1-203. [ Links ]
45. Atkinson R. Atmospheric chemistry of VOCs and NOx. Atmos Environ. 2000;34:2063-2101. [ Links ]
46. Gertler AW, Miller DF, Lamb D, Katz U. Studies of sulphur dioxide and nitrogen dioxide reactions in haze and cloud. In: Durham JL, editor. Chemistry of particles, fogs and rain, Acid Precipitation Series vol. 2. Boston: Butterworth, 1984; p. 131-160. [ Links ]
47. Cardoso AA, Liu H, Dasgupta PK. Fluorometric fiber optic drop sensor for atmospheric hydrogen sulphide. Talanta. 1997;44:1099-1106. [ Links ]
48. Blanchard CL, Michaels H, Tannenbaum S. Regional estimates of acid deposition in fluxes in California 1985-1994. Sacramento: Californian Air Resources Board; 1996. [ Links ]
49. Weiss SB. Cars, cows, and checkerspot butterflies: Nitrogen deposition and management of nutrient-poor grasslands for a threatened species. Conserv Biol. 1999;13(6):1476-1486. [ Links ]
50. Fenn ME, Haeuber R, Tonnesen GS, et al. Nitrogen emissions, deposition and monitoring in the western United States. Bioscience. 2003;53(4):391-403. [ Links ]
51. Stevens CJ, Dise NB, Mountford JO, Cowing DJ. Impact of nitrogen deposition on the species richness of grasslands. Science. 2004;303(5665):1876-1879. [ Links ]
52. Nilsson S, Grennfelt P. Critical loads for sulphur and nitrogen. Nord miljorapport 15. Report from a workshop held at: Skokloster, Sweden, 1988 Mar 19-24. Copenhagen: Nordic Council of Ministers; 1988. p. 225-268. [ Links ]
53. Grennfelt P, Thörnelöf E. Critical loads for nitrogen. Nord miljorapport 41. Report from a workshop held at: Lokeberg, Sweden, 1992 Apr 6-10. Copenhagen: Nordic Council of Ministers; 1992. [ Links ]
54. Josipovic M, Annegarn HJ, Kneen MA, Pienaar JJ, Piketh SJ. Concentrations, distributions and critical level exceedance assessment of SO2, NO2 and O3 in South Africa. Environ Monit Assess. 2009, doi: 10.1007/s10661-009-1270-5. [ Links ]
 Correspondence to:
Correspondence to:
Kirsten Collett
Postal address:
Climatology Research Group, University of the Witwatersrand
Private Bag X3, WITS
2050, South Africa
email: kirsten@crg.wits.ac.za
Received: 10 Sept. 2009
Accepted: 18 Feb. 2010
Published: 01 June 2010
This article is available at: http://www.sajs.co.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}