










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Science
versión On-line ISSN 1996-7489
versión impresa ISSN 0038-2353
S. Afr. j. sci. vol.106 no.1-2 Pretoria ene./feb. 2010
RESEARCH LETTER
The effects of Sutherlandia frutescens extracts in cultured renal proximal and distal tubule epithelial cells
Alisa Phulukdaree; Devapregasan Moodley; Anil A. Chuturgoon
Discipline of Medical Biochemistry, School of Medical Sciences, Faculty of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
ABSTRACT
Sutherlandia frutescens (SF), a medicinal plant indigenous to South Africa, is traditionally used to treat a diverse range of illnesses, including cancer and viral infections. The biologically active compounds of SF are polar, thus renal elimination increases susceptibility to toxicity in that organ. This study investigated the antioxidant potential, lipid peroxidation, mitochondrial membrane potential and apoptotic induction by SF extracts on proximal and distal tubule epithelial cells. Cell viability was determined using the MTT assay. Mitochondrial membrane potential was determined using a flow cytometric JC-1 Mitoscreen assay. Cellular glutathione and apoptosis were measured using the GSH-GloTM Glutathione assay and Caspase-Glo® 3/7 assay, respectively. The IC50 values from the cell viability results for LLC-PK1 and MDBK were 15 mg/mL and 7 mg/mL, respectively. SF extracts significantly decreased intracellular glutathione in LLC-PK1 (p < 0.0001) and MDBK (p < 0.0001) cells, while lipid peroxidation increased in treated LLC-PK1 (p < 0.0001) and MDBK (p < 0.0001) cells. JC-1 analysis showed that SF extracts promoted mitochondrial membrane depolarization in both LLC-PK1 and MDBK cells by up to 80% (p < 0.0001). The activity of caspase 3/7 increased in both LLC-PK1 (11.9-fold; p < 0.0001) and MDBK (2.2-fold; p < 0.0001) cells. SF extracts at high concentrations appear to increase oxidative stress, to alter mitochondrial membrane integrity, and to promote apoptosis in renal tubule epithelia.
Keywords: Sutherlandia frutescens; antioxidant; lipid peroxidation; mitochondrial depolarisation; apoptosis
INTRODUCTION
Sutherlandia frutescens (SF), a member of the Leguminosae family, is a multipurpose medicinal plant endemic to South Africa.1 Commonly known as 'cancer bush', it has been used in crude form for years by traditional healers to treat a variety of ailments including internal cancers, diabetes, uterine disease, influenza, HIV, depression, and arthritis. 1 Various doses of SF leaf powder have been administered to humans, but have produced no known side effects.
Leaves of SF contain the biologically active compounds L-canavanine, D-pinitol, gamma amino butyric acid (GABA), parabens, saponins, cycloartane glycosides and triterpenoid diglucoside.2,3,4 L-canavanine stores nitrogen in seeds and is used in plant chemical defense mechanisms.5,6 Its production by SF is dependent on the availability of abiotic factors.7 L-canavinine, a non-protein amino acid, is a structural analogue of L-arginine. It can be recognised by arginine-utilising enzymes such as arginyl-tRNA synthetase, and consequently can be incorporated into newly synthesised peptides.5 Canaline, produced by arginase-mediated hydrolytic cleavage of L-canavanine, has been shown to have anti-tumour properties.8 D-pinitol, a chiro-inositol sugar, possesses anti-diabetic properties and is used in the treatment of wasting in cancer and HIV/AIDS patients.9,10
GABA, an inhibitory neurotransmitter, mediates most of its effects inside the nervous system.11 It has been used as a drug for the relief of anxiety and stress.12 Interestingly, GABA has been shown to affect the absorption of ions in the renal tubules.13 Intracellularly, GABA is metabolised through the action of the enzymes glutamate decarboxylase, GABA transaminase and succinic semialdehyde dehydrogenase, and is transformed into citric acid cycle intermediates.14
Many experiments have been done to determine the mechanism(s) of action of SF extracts. In an in vivo experiment it was concluded that SF extracts possessed anticonvulsant effects in mice subjected to the induction of epilepsy.15 SF-treated human breast adenocarcinoma cells in culture showed morphological characteristics of apoptosis and cell growth inhibition.16,17 An antiproliferative effect of SF was demonstrated to be concentration-dependent in breast cancer and leukaemia cell lines, with no significant antioxidant effects.2 Another study, by contrast, showed that SF extracts displayed antioxidant potential (hydroxyl free radical and superoxide scavenging properties) in cell-free and stimulated neutrophil systems.18 An apparent antiretroviral activity of SF has been thought to be mediated by the inhibition of HIV-1 target enzymes, such as HIV-1 reverse transcriptase.19
The route of elimination of polar compounds from the circulation occurs via the renal system. The kidney functions to filter blood, allowing substances to enter Bowman's capsule and the renal tubules. Filtered nutrients are actively reabsorbed at the proximal convoluted tubule (PCT), while ions are actively reabsorbed at the distal convoluted tubule (DCT).20 Based on their contrasting functions, the PCT and DCT epithelia have different cell architectures.21
The PCT epithelium has a brush border of tall microvilli that extends into the lumen to increase the surface area 20-fold for the efficient reabsorption of molecules from the glomerular filtrate back into circulation. Histologically, PCT cells stain intensely with eosin due to their high content of organelles and mitochondria. The PCT is responsible for the active reabsorption of 99% of glucose and amino acids from the glomerular filtrate.21 The DCT cells are smaller, simple cuboidal epithelial cells (stain less intensely due to fewer organelles) that actively reabsorb sodium from the tubular fluid.21 The close proximity of filtered substances to this kind of tubular epithelium increases the susceptibility of these cells to damage.
To date, limited scientific evidence has been available on the mechanism by which SF extracts affect cellular processes and the side effects related to their use. This medicinal plant, however, continues to be recommended as a traditional remedy and is used by a large portion of the South African community. In this study, the nephrotoxic and apoptotic effects of SF extracts on two kidney cell lines, LLC-PK1 (PCT epithelium) and MDBK (DCT epithelium), were investigated and compared.
MATERIALS AND METHODS
Materials
Sutherlandia frutescens tablets (Phyto NovaTM, Cape Town, South Africa) were purchased from a local pharmacy. The LLC-PK1 and MDBK cell lines were purchased from Highveld Biologicals (Johannesburg, South Africa). All tissue culture reagents, the GSH-GloTM Glutathione Assay and the Caspase-Glo® 3/7 Assay were obtained from Whitehead Scientific (Johannesburg, South Africa). The JC-1 dye was purchased from BD Biosciences (South Africa). All other reagents were purchased from Merck (South Africa) unless otherwise stated.
Preparation of Sutherlandia frutescens extracts
Phyto Nova SutherlandiaTM tablets were used to prepare an aqueous extract of the active ingredients of the plant. Sixty tablets were crushed to a fine powder in a pestle and mortar, weighed and suspended in deionised water (1.2 g per 10 mL). The mixture was continuously stirred at room temperature for 1.5 h, and thereafter transferred to 50 mL sterilin tubes and centrifuged (3 645 g, 10 min) at room temperature. The upper aqueous layer (SF extract) was removed, vacuum filtered and stored at 4 ºC. SF extract dilutions (24 mg/mL, 12 mg/mL, 6 mg/mL, 2.4 mg/mL, 1.2 mg/mL, 0.6 mg/mL, and 0.3 mg/mL) were prepared using complete culture media (CCM), comprising Eagle's minimum essential medium, 10% foetal calf serum, 1% L-glutamine and 1% penstrepfungizone.
Cell culture and cytotoxicity assay
LLC-PK1 and MDBK cells were cultured (37 ºC, 5% CO2) to confluency in 75 cm3 flasks in CCM. The cytotoxicity of SF in LLC-PK1 and MDBK cells was measured using the MTT assay.22 LLC-PK1 and MDBK cells (10 000/well) were incubated with varying SF extract dilutions for 48 h in triplicate in microtitre plates, together with the respective controls (cells incubated with CCM only). The cells were then incubated with the MTT substrate (5 mg/mL) for 4 h. Thereafter all supernatants were aspirated, and dimethyl sulphoxide (DMSO) (100 µL/well) was added to the wells. Finally, the optical density was measured at 570 nm, with a reference wavelength of 690 nm, by an enzyme-linked immunosorbent assay (ELISA) plate reader (Bio-Tek µQuant). The data were translated to 'percentage cell viability' versus 'concentration of extract', from which the IC50 (half the maximal inhibitory concentration) values for each cell line and for the combination of treatments were determined. For all subsequent biochemical assays, both cell lines were grown to confluency and treated with the determined IC50 values of the SF extracts.
Lipid peroxidation assay
Oxidative damage of both cell lines was assessed using the thiobarbituric acid assay, because lipid peroxidation is commonly quantified by levels of malondialdehyde (MDA). After the 48 h incubation with SF extract, the culture fluid from each SF-treated flask and from the untreated controls (500 µL) was dispensed into duplicate glass tubes (one representing the sample, one representing a negative control), followed by addition of 7% H2PO3 (200 µL). A positive control of 1% MDA was prepared. Thiobarbituric acid (1%, w/v)/ 0.1 mM butylated hydroxytoluene solution (400 µL) was added to sample tubes. To the tubes with the negative controls, 400 µL of 3 mM HCl was added. The solution was adjusted to pH 1.5 and heated at 100 C for 15 min. Once cooled, butanol (1.5 mL) was added and the sample then centrifuged (8 400 g, 6 min). Following centrifugation, the butanol phase (200 µL) from each sample and from the blanks was aliquoted into a microtitre plate. The optical density was measured at 532 nm, with a reference wavelength of 600 nm, by an ELISA plate reader. The sample means of ten replicates were calculated and divided by the absorption coefficient, 156 mM-1.
Glutathione assay
The GSH-GloTM Glutathione Assay (Promega, Madison, USA) was used to measure glutathione (GSH) levels. Cells (those treated with SF extract and the untreated controls after 48 h incubation) were transferred to an opaque microtitre plate (50 µL of 10 000 cells/well, 10 replicates). GSH standards (0 µM - 5 µM) were prepared from a 5 mM stock solution diluted in water. Five two-fold dilutions of the GSH stock were prepared and transferred into wells (50 µL) of the microtitre plate. The 2X GSH-GloTM Reagent was prepared according to the manufacturer's instructions, added to the experimental wells (50 µL/well), and incubated at room temperature. Reconstituted Luciferin Detection Reagent (50 µL) was added to each well and incubated. The luminescence was measured on a ModulusTM microplate luminometer (Turner Biosystems, Sunnyvale, USA). A standard curve was derived using the GSH standards (0 µM - 5 µM) and the GSH concentration in each sample was extrapolated from the equation.
Caspase-3/7 assay
The apoptotic potential of SF extracts on both cell lines was determined using the Caspase-Glo®3/7 assay (Promega). Caspase-Glo®3/7 Reagent was reconstituted according to the manufacturer's instructions and added to both the SF-treated and control cells (following 48 h incubation) in the wells of a microtitre plate (10 µL reagent per 50 µL of 10 000 cells/well, 10 replicates) and incubated in the dark (30 min). The luminescence was measured on a ModulusTM microplate luminometer (Turner BioSystems). The caspase-3/7 activity of the SF-treated samples was represented as X-fold change compared to the control (cells incubated with CCM only).
Mitochondrial membrane potential
The mitochondrial membrane potential (ΔΨm) of LLC-PK1 and MDBK cells (both those treated with SF extract and their untreated controls; 48 h) was assessed using fluorescence-activated cell sorting (FACS) and the JC-1 Mitoscreen assay (BD Biosciences) according to the manufacturer's instructions. Cells (approximately 100 000) were transferred to polystyrene cytometry tubes. The JC-1 dye (150 µL) was added to the cells and allowed to incubate (37 ºC, 5% CO2, 10 min). Cells were then washed with JC-1 Mitoscreen wash buffer (400 g, 5 min) and resuspended in 300 µL flow cytometry sheath fluid. Flow cytometry data from stained cells (15 000 events) was obtained using a FACSCalibur (BD Biosciences) flow cytometer with CellQuest PRO v4.02 software (BD Biosciences). Cells were gated to exclude debris using FlowJo v7.1 software (Tree Star Inc., Ashland, USA)
Statistical analysis
Results are expressed as the means, with error bars representing the standard deviations (s.d.) of the means. Statistical significance between samples for the MTT assay was determined using the one-way ANOVA Tukey Kramer Multiple Comparisons test. The statistical significances of the lipid peroxidation assay, GSH assay, caspase-3/7 assay and mitochondrial depolarisation assay were determined by the Mann-Whitney test for non-parametric data using GraphPad InStat Software, (GraphPad Software Inc., La Jolla, USA).
RESULTS
Cytotoxicity
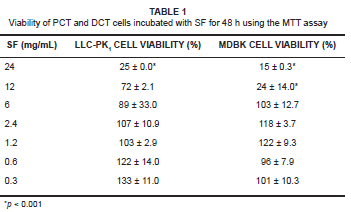
The cytotoxic effects of SF extracts (MTT assay) were determined in both LLC-PK1 and MDBK cells treated with a range of extract dilutions for 48 h (see Table 1). The IC50 for SF extract was determined as 15 mg/mL and 7 mg/mL dilutions for the LLC-PK1 and MDBK cells, respectively. The cell viability of both cell lines treated with concentrations between 6 mg/mL and 0.3 mg/mL was more than 89%. However, at higher SF extract concentrations of 24 mg/mL and 12 mg/mL, respectively, cell viability was decreased to 25% and 72% of controls (p < 0.001) in the case of LLC-PK1 cells' and to 15% and 24% (p < 0.001) of controls in the case of MDBK cells.
GSH assays
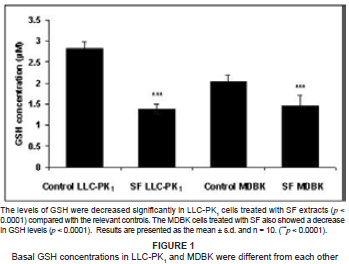
The intracellular concentrations of GSH in both SF-extract-treated renal epithelial cell lines were determined. They decreased significantly in SF-treated LLC-PK1 cells as compared with controls (p < 0.0001), whilst there also was a significant decrease in GSH in SF-treated MDBK cells as compared with controls (p < 0.0001) (see Figure 1).
Lipid peroxidation assays
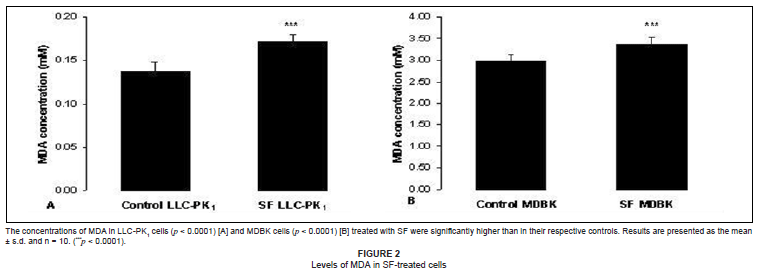
In order to determine whether mitochondrial damage and cytotoxicity were related to oxidative stress, we measured the levels of lipid oxidation products in both SF-treated cell lines. There were significantly higher levels (p < 0.0001) of MDA in both the SF-treated cell lines as compared with the respective control cells (see Figures 2A and 2B).
Mitochondrial membrane potential analyses
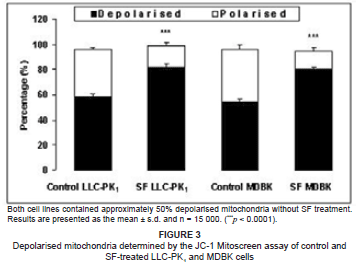
To determine whether LLC-PK1 and MDBK cells were metabolically viable after SF-extract treatment, we investigated changes in ΔΨm. The SF-treated LLC-PK1 cells had a significantly higher (p < 0.0001) percentage of depolarised mitochondria as compared with the untreated cells (80.2% vs 54.6%) (see Figure 3). Similarly, the MDBK cells had a significantly higher (p < 0.0001) percentage of depolarised mitochondria as compared with controls (81.7% vs 58.2%) (see Figure 3).
Caspase-3/7 assay
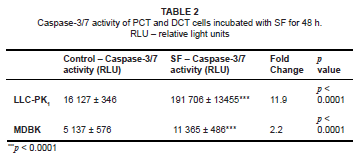
A significant change in ΔΨm is a good indicator of apoptosis. The intracellular activities of caspase 3/7 in both SF-treated cell lines were measured (as a fold-change in comparison with the relevant controls). The activity of caspase 3/7 in SF-treated LLC-PK1 cells showed a highly significant 11.9-fold increase (p < 0.0001) compared with the controls. The SF-treated MDBK cells also showed a significant 2.2-fold increase (p < 0.0001) over controls (see Table 2). These results indicated that PCT cells were more susceptible to apoptotic induction by SF than were DCT cells.
DISCUSSION
SF is used by many South Africans as a traditional remedy or ameliorant for many diseases, including HIV infection. The crude plant concoction is normally taken orally and the main route of elimination of the absorbed constituent polar compounds is via the renal system. The potential for injury by noxious compounds in the PCT region is high due to the ability of the PCT cells to concentrate substances that have been filtered by the glomerulus.20
The results of this investigation have shown that both kinds of renal tubular cells, LLC-PK1 and MDBK cells, are affected by SF extracts in vitro. This sensitivity may be attributed to the location and functional capacities of these cells. The intact PCT is susceptible to injury, as it is at this site of the nephron where toxicants accumulate and where there is an abundance of mitochondria for active re-absorption and transportation of ions, low molecular weight proteins, glutathione conjugates and heavy metals.20 Cells treated with SF extracts in vitro, at concentrations of 6 mg/mL and lower, displayed greater than 89% viability. At such low concentrations, SF extracts appeared to promote cell metabolism as evidenced by the increased conversion of the MTT salt. This might have been due to both increased mitochondrial reductase enzyme activity,22 and increased availability of reducing equivalents such as NADH, a byproduct of metabolic pathways such as glycolysis and the Krebs cycle.23 The anti-diabetic and favourable cellular metabolic properties of SF were previously demonstrated in male Wistar rats.24
The hypoglycaemic potential of SF was shown in diabetogenic rats, where it exhibited properties similar to those of the extracts similar to those of metformin, the common diabetes type-2 drug.24 SF extracts increased the uptake of glucose by the kidney and resulted in increased glycolysis, which increased the rate of the Krebs cycle and produced more reducing equivalents (NADH and FADH2). The high concentration of reducing equivalents donate electrons to the electron transport chain (ETC), resulting in increased ATP production via oxidative phosphorylation. This increased ATP concentration, is utilised for the active reabsorption of substances by the PCT and DCT. The continuous flux of electrons along the ETC and pumping of protons out of the matrix results in a negative mitochondrial membrane potential, during which process reactive oxygen species (ROS) are generated as a byproduct, rendering the mitochondria susceptible to oxidative damage.25
Free radicals attack polyunsaturated fatty acids, a major component of cell membranes, thereby forming lipid peroxyl radicals. Lipid peroxidation is initiated when the reactive lipid peroxyl radicals are not detoxified by antioxidants. The lipid alkoxyl radical undergoes cyclisation forming an intermediate product which can degrade into MDA.26 Lipid peroxides can be detoxified by conjugation to antioxidant molecules such as ascorbic acid, α-tocopherol, uric acid and GSH.27 Glutathione concentrations were significantly decreased in SF-treated LLC-PK1 and MDBK cells with a corresponding increase in lipid peroxidation compared to the controls (p < 0.0001).
Both the LLC-PK1 and MDBK cells were transformed cell lines which proliferated rapidly and possessed high metabolic activity which resulted in increased production of ROS, which, when not sequestered rapidly, increased lipid peroxidation. The MDA concentrations in the control LLC-PK1 and MDBK cells were 0.14 mM and 3.0 mM, respectively (see Figures 2A and 2B), whereas the concentrations of GSH in LLC-PK1 and MDBK cells were 2.85 µM and 2.1 µM. respectively (see Figure 1). The increased basal GSH concentration in the LLC-PK1 cells suggested that these cells were better protected from increased ROS production and subsequent peroxidation than the MDBK cells were.
The oxidative ability of L-canavanine to induce ROS and ensuing lipid peroxidation was determined previously in a mouse glial cell line (N11)27. In this study, a luminol-amplified chemiluminescence assay showed that 1 mM of L-canavanine significantly (p < 0.05) increased ROS production and lipid peroxidation in N11 cells.27 L-canavanine, a structural analogue of L-arginine, is a competitive inhibitor of arginine-utilising enzymes such as nitric oxide (NO) synthase, which synthesises NO from L-arginine.28 Nitric oxide is involved in many biological processes and has the ability to spontaneously react with superoxide radicals to produce peroxynitrite and subsequently, nitrate.27 Nitric oxide, therefore, serves as a reactive radical scavenger that ultimately prevents oxidative damage to cellular components.29 The possible decreased activity of NO synthase and inhibition of NO synthesis by SF may explain the significant lipid peroxidation in these renal cell lines.
Another possibility for the increased lipid peroxidation in both the SF-treated LLC-PK1 and MDBK cells may be due to a decreased ΔΨm. GABA was shown to penetrate up to 60% of the mitochondrial matrix volume in rat brain and liver cells. The weak acidic nature of GABA may contribute to the proton gradient disruption within mitochondria.30 A change in ΔΨm promotes formation of ROS as it offsets the normal functioning of the ETC.
In addition to its acidic nature, high concentrations of GABA cause the reduction of intra-mitochondrial NAD+ stimulated by glutamate.30 Excessive intra-mitochondrial NADH is dissipated by donating its electrons to the ETC. The metabolism of GABA involves the enzymes GABA: pyruvate transaminase, GABA: α-ketogluterate transaminase and succinic semialdehyde dehydrogenase to form succinate which can enter the Krebs cycle and produce substrates for the ETC.14
ROS production in the mitochondria increases susceptibility for membrane damage, altering the mitochondrial permeability transition. This may account for the high percentage of depolarised mitochondria noted after treatment of cells with SF extracts. The alteration of the ΔΨm within a cell will not, however, occur simultaneously to all the mitochondria that are present.31 The number of mitochondria that are affected determines the amount of ATP generated and the ultimate fate of the cell. The amount of ATP available will determine the type of cell death that occurs within the cell.20
When mitochondrial membranes are depolarised, pro-apoptotic signals are released. It was demonstrated that changes in ΔΨm favoured the transition of a tightly bound form of cytochrome c (to cardiolipin in the inner mitochondrial membrane) into its loosely bound form, followed by the release of cytochrome c into the extra-mitochondrial environment.32 The release of cytochrome c, Apaf-1 and calcium ions into the cytosol results in the activation of downstream caspases that execute apoptosis.25,33,34 This study showed that SF-extract treatments increased caspase-3/7 activity in both cell lines (by 11.9-fold in LLC-PK1cells and by 2.2-fold in MDBK cells).
When comparing the change in ΔΨm of LLC-PK1 and MDBK cells with caspase-3/7 activity, it became evident that SF extracts are potent apoptotic inducers. The percentage mitochondrial membrane depolarisation in both cell lines appears to be equal at basal levels; following SF-extract treatments, an almost similar increase in depolarisation occurs in both LLC-PK1 and MDBK cells.
The 11.9-fold increase of caspase activity in LLC-PK1, as compared to a 2.2-fold increase in MDBK cells, may be due to the difference in mitochondrial numbers and cellular architecture of both cell lines. It is known that PCT epithelium contains more mitochondria than do DCT cells. The depolarisation of mitochondria allows for the increased release of more pro-apoptotic signals, thereby resulting in increased apoptosis.
There exists a real concern about the unregulated use of the crude plant derivatives of SF. Evidence that the recommended dosages are in such concentrations that they may be potentially harmful to the renal tubular cells, could be attained in vivo. Ojewole determined that SF extract had an LD50 of 1 280 mg/kg body weight in rats.35 In a clinical trial on ten healthy adults receiving a dose of 800 mg/day of SF extract, no significant changes in haematological, physiological and biochemical markers were noted after three months of treatment.36 The only changes observed in this in vivo study were a decrease in respiratory rate (p < 0.04), and increases in platelet count (p < 0.05), mean cell haemoglobin (p = 0.01), protein (p < 0.03) and albumin levels (p < 0.03).36 Chronic doses of SF extract, however, may eventually lead to renal toxicity.
CONCLUSION
The aqueous SF extracts studied here were not cytotoxic at low concentrations, but had the potential to increase oxidative stress, alter the integrity of mitochondrial membranes, and promote apoptosis in renal tubule epithelia at high concentrations in vitro.
ACKNOWLEDGEMENTS
Miss A. Phulukdaree thanks the University of KwaZulu-Natal's LEAP Mellon Foundation for a scholarship.
REFERENCES
1. Gericke N, Albrecht CF, Van Wyke B, Mayeng B, Mutwa C, Hutchings A. Sutherlandia frutescens. Am J Mens Health. 2001;13:9-15. [ Links ]
2. Tai J, Cheung S, Chan E, Hasman D. In vitro culture studies of Sutherlandia frutescens on human tumour cell lines. J Ethnopharm. 2004;9:9-19. [ Links ]
3. Fu X, Li X-C, Smillie TJ, et al. J Nat Prod. 2008;71:1749-1753. [ Links ]
4. Olivier DK, Albrecht CF, van Wyk B-E, van Heerden F. SU3, an oxocycloartane diglucoside from Sutherlandia humilis. Phytochem Lett. 2009;2:123-125. [ Links ]
5. Bence AK, Crooks PA. The Mechanism of L-canavanine cytotoxicity: arginyl tRNA synthetase as a novel target for anticancer drug discovery. J Enzym Inhib Med Chem. 2003;18:383-394. [ Links ]
6. Rosenthal GA. The biological effects and mode of action of L-canavanine, a structural analogue of L-arginine. Q Rev Biol. 1977;52:155-178. [ Links ]
7. Colling J, Kossman J, Makunga NP. Piecing together Sutherlandia frutescens (L.) R. Br. metabolism using in vitro tools. S Afr J Bot. 2009;75:396. [ Links ]
8. Crooks PA, Rosenthal GA. Use of L-canavanine as a chemotherapeutic agent for the treatment of pancreatic cancer. US Patent. 1994;5:552,440. [ Links ]
9. Bates SH, Jones RB, Bailey CJ. Insulin-like effect of pinitol. Br J Psychiatry. 2000;130:1944-1948. [ Links ]
10. Ostlund RE, Sherman WR. Pinitol and derivatives thereof for the treatment of metabolic disorders. US Patent. 1996;5:827,896. [ Links ]
11. Van Wyk BE. A broad review of commercially important Southern African medicinal plants. J Ethnopharm. 2008;119:342-355. [ Links ]
12. Sia, C. Spotlight on ehnomedicine: Usability of Sutherlandia frutescens in the treatment of diabetes. RDS. 2004;1:145-149. [ Links ]
13. Parducz A, Dobo E, Joachim RW, Petrusz P, Erdo SL. GABA-immunoreactive structures in rat kidneys. J Histochem Cytochem. 1992;40:675-680. [ Links ]
14. Shelp BJ, Bown AW, McLean MD. Metabolism and functions of gamma-aminobutyric acid. Trends Plant Sci. 1999;4:446-452. [ Links ]
15. Ojewole JAO. Anticonvulsant property of Sutherlandia frutescens R. BR. (variety Icnana E. MEY.) [Fabraceae] shoot aqueous extract. Brain Res Bull. 2008;75:126-132. [ Links ]
16. Stander BA, Marais S, Steynberg TJ, et al. Influence of Sutherlandia frutescens extracts on cell numbers, morphology and gene expression in MCF-7 cells. J Ethnopharm. 2007;112:312-318. [ Links ]
17. Stander A, Marias S, Stivaktas V, et al. In vitro effects of Sutherlandia frutescens water extracts on cell numbers, morphology, cell cycle progression and cell death in a tumorigenic and a non-tumorigenic epithelial breast cell line. J Ethnopharm. 2009;124:45-60. [ Links ]
18. Fernandes AC, Cromarty AD, Albrecht C, Van Rensburg JEC. The antioxidant potential of Sutherlandia frutescens. J Ethnopharm. 2004;95:1-5. [ Links ]
19. Harnett SM, Oosthuizen V, Van de Venter M. Anti-HIV activities of organic and aqueous extracts of Sutherlandia frutescens and Lobostemon trigonus. J Ethnopharm. 2005;96:113-119. [ Links ]
20. Klaassen CD. Cassarett and Doull's Toxicology, The basic science of poisons, 6th ed. Columbus: McGraw Hill, 2001; p. 596-600. [ Links ]
21. Young B, Lowe JS, Stevens A, Heath JW. Wheater's functional histology: a text and colour atlas. 4th ed. University of Michigan: Churchill Livingstone. 2000; p. 268-273. [ Links ]
22. Mossman T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65:55-63. [ Links ]
23. Berridge MV, Tan AS, Herst PM. Tetrazolium dyes as tools in Cell Biology: new insights into their cellular reduction. Biotechnol Annu Rev. 2005;11:127-152. [ Links ]
24. Chadwick WA, Roux S, Van de Venter M, Louw J, Oelofsen W. Anti-diabetic effects of Sutherlandia frutescens in Wistar rats fed a diabetogenic diet. J Ethnopharm. 2006;109:121-127. [ Links ]
25. Waterhouse NJ, Ricci J, Green DR. And all of a sudden it's over: mitochondrial outer-membrane permeabilization in apoptosis. Biochimie. 2002;84:113-121. [ Links ]
26. Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57:715-725. [ Links ]
27. Riganti C, Aldieri E, Bergandi L, et al. Nitroarginine methyl ester and canavanine lower intracellular reduced glutathione. Free Radic Biol Med. 2003;35:1210-1216. [ Links ]
28. Luzzi SD, Marletta MA. L-Arginine analogs as alternate substrates for nitric oxide synthase. Bioorg Med Chem Lett. 2005;15:3934-3941. [ Links ]
29. Li CQ, Wogan GN. Nitric oxide as a modulator of apoptosis. Canc Lett. 2005;226:1-15. [ Links ]
30. Brand MD, Chappell JB. Permeability of mitochondria from rat liver and rat brain to GABA. J Neurochem. 1973;22:47-51. [ Links ]
31. Lemasters JJ, Nieminen AL, Qian T. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim, Biophys Acta. 1998;1366:177-196. [ Links ]
32. Ott M, Robertson JG, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259-1263. [ Links ]
33. Lalier L, Cartron P-F, Juin P, et al. Bax activation and mitochondrial insertion during apoptosis. Apoptosis. 2007;12:887-896. [ Links ]
34. Ott M, Gogvadze V, Orrenius S, Zhitvotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913-922. [ Links ]
35. Ojewole JAO. Analgesic, anti-inflammatory and hypoglycemic effects of Sutherlandia frutescens R.Br. (variety incana E.MEY.) [Fabaceae] shoot aqueous extract. Methods and findings in experimental and clinical pharmacology, 2004;26(6):409-416. [ Links ]
36. Johnson Q, Syce J, Nell H, Rudeen K, Folk WR. A randomized, double-blind, placebo-controlled trial of lessertia frutescens in healthy adults. PLOS Clinical Trials. 2007;16:0001-0007. [ Links ]
 Correspondence to:
Correspondence to:
Anil Chuturgoon
Discipline of Medical Biochemistry, School of Medical Sciences
University of KwaZulu-Natal, Private Bag 7
Congella 4013, Durban, South Africa
email: chutur@ukzn.ac.za
Received: 03 Sept. 2009
Accepted: 08 Dec. 2009
Published: 11 Mar. 2010
This article is available at: http://www.sajs.co.za


{kind=link}