










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.105 n.7-8 Pretoria Jul./Aug. 2009
RESEARCH ARTICLES
The influence of cation, anion and water content on the rate of formation and pore size distribution of zeolite ZSM-5
L. Petrik
Environmental and Nano Sciences, Department of Chemistry, University of the Western Cape, Private Bag X17, Bellville 7535, South Africa. E-mail: lpetrik@uwc.ac.za
ABSTRACT
Rapid synthesis and high mesoporosity of ZSM-5 is promoted by altering a fixed molar regime with respect to anion and cation type and water content only. This study shows that merely changing the cation and anion types, accompanying the hydroxide and alumina (Al) sources, respectively, has an impact on the characteristics of ZSM-5. Under identical and replicated preparation and testing conditions, the synthesis time, Al incorporation, product yield, acidity, morphology, pore size distribution and catalytic activity were affected. An ion pair combination of Na+/NO3– allowed the formation of ZSM-5 with enhanced mesoporosity within 3 h in a high-water environment, whereas ZSM-5 prepared with the Na+/SO42– combination had enhanced n-hexane cracking activity. Use of a K+/NO3– ion pair combination in a high-water environment resulted in slower time of formation and larger ZSM-5 crystals with extra-framework Al, loss of acidity, lower surface areas and reduced n-hexane cracking activity. The sulphate anion inhibited the incorporation of Al in ZSM-5 products in low-water environments. Low-water levels increased the time of formation of ZSM-5.
Key words: zeolite ZMS-5, synthesis, cation, anion, acidity, porosity, extra-framework aluminium, n-hexane cracking activity, sulphate, nitrate
Introduction
Apart from microwave-assisted techniques,1 few studies report or give a rationale for rapid crystallisation of zeolites. Inui2 reported a method for rapid synthesis of zeolite ZSM-5 (MFI-type zeolite according to IUPAC nomenclature) using an involved pre-synthesis preparation technique, in which filtered, ball-milled, aged precursor gels containing high amounts of NaCl were reintroduced into fresh mother liquors and subjected to hydrothermal synthesis for 0.5 h.
Furthermore, few investigations were carried out in comparable molar regimes in systematic studies concerning the influence of cation and anion type on the rate of formation, morphology and catalytic activity of ZSM-5. Most investigations report possible cation and anion effects in terms of salt additions, thus inevitably introducing changes in molar concentrations of reactant species and hence not keeping all extraneous variables constant.3–7 Accompanying anions are treated as spectator anions and levels are not controlled. Thus it is difficult to decouple the relative effects of each variable on catalyst quality, or to correlate changes in catalytic activity of ZSM-5 with synthesis variables, when molar regimes and sources of chemicals varied from study to study. Different preparation methods for ZSM-5 may result in the introduction of diffusional constraints and changes in shape-selectivity of the working catalyst due to changes such as the accidental introduction of extra-framework aluminium (EFAl), or altered crystal morphology, pore volume distribution and surface areas. Thus different activities reported, when comparing the catalytic activity of ZSM-5 for a particular reaction, may be caused by different preparation conditions for this zeolite type between studies.
Petrik et al.8 investigated the influence of physical and chemical parameters on the properties of ZSM-5 by using a molar regime which allowed relatively rapid crystallisation. In this study, the effect of changing the cation and anion types accompanying the hydroxide and alumina source, on synthesis kinetics, pore volume distribution, acidity, morphology and catalytic activity was investigated in a fixed molar regime, using standard hydrothermal preparation conditions. The effect of high and low water content was also noted. n-Hexane cracking activity9 was used as a probe reaction to determine the relative acidity and catalytic activity of several of the variously prepared ZSM-5 catalysts.
Methods
To investigate the influence of the cation and anion type and water concentration on product quality of ZSM-5 in a carefully controlled environment, four different ion pair combinations were used during hydrothermal synthesis, namely: Na+/SO42–; K+/SO42–; Na+/NO3– and K+/NO3–. The molar composition and species type are shown in Table 1. The source of hydroxide was either NaOH or KOH, the alumina source was either Al2(SO4)318H2O or Al(NO3)39H2O·SiO2 supplied by use of the colloidal silicon source, Ludox AS 40, and the template used was tetrapropylammonium bromide (TPABr). Each combination was tested in a high-water and low-water environment and all syntheses were replicated, using the two different molar quantities of water, as specified in Table 1. To eliminate extraneous variables and allow comparison, the molar concentration of the cation (OH–:Si:H2O:TPABr:Al) was kept rigorously constant, except that, due to stoichiometric constraints, 3 mols of NO3– were provided per mol aluminium, whereas, in the case of the SO42– combinations, only 1.5 mols of SO42– per mol aluminium were provided. A compromise had to be made between the importance of a constant Si/Al ratio in terms of catalytic activity, and the constant anion quantity in terms of the influence of the type of anion on the catalytic properties. It is impossible to keep anion concentration exactly the same when different sources of Al are used. Hence the differences observed between the effects of the NO3– compared to the SO42– anion may be partially due to the relative concentrations of each anion. Reactants were chosen in such a way that the amount of hydroxide was kept constant, to exclude the influence of changes in hydroxide concentration on product characteristics.
The silica source used was Ludox AS 40 commercial silica from Du Pont. The Al source used was either aluminium sulphate by Merck with a purity of 99%, or aluminium nitrate by Saarchem, with a purity of 99%. The organic templating agent was tetrapropylammonium bromide (TPABr) from Riedel de Häen (purity of >99%). The sodium hydroxide (Saarchem) used in this study had a purity of 97% whereas the potassium hydroxide (Saarchem) had a purity of 85% (>12% H2O, 2% K2CO3 < 0.004% impurities such as Pb, Fe, As, N, Cl).
During the pre-synthesis preparation stage, the reagents were added together in the following sequence, taking rigorous care to be exact. A third of the amount of water was used to dissolve the TPABr in a 200-ml glass beaker while stirring magnetically. The Al source was added to the required dry mass of NaOH and dissolved in about 5–10 ml of the deionised water by heating to boiling point. The mass of water lost due to evaporation during heating was adjusted by the addition of an equivalent mass of deionised water. This alkaline solution was added slowly to the aqueous template solution and traces of reactant solutions on the glassware were rinsed with the remaining deionised water. The Ludox was added dropwise under strong agitation by use of magnetic stirring. The mixture was vigorously stirred for about 10 min in all cases. About 12 g of the pre-synthesis gel mixture was transferred into the thoroughly cleaned Teflon liners. The zeolite synthesis was carried out statically in Teflon-lined carbon steel Parr Bombs® with a volume of 30 ml at autogeneous pressure. The autoclaves were loaded to a capacity of 40% with a portion of the respective pre-synthesis gel mixture, which corresponded to a weight of about 12 g of the pre-synthesis gel mixture, replicated in each case. These reaction vessels were placed in a preheated hot-air oven at 160°C. All hydrothermal syntheses were performed statically, in order to obtain a time of formation which would not be influenced by secondary nucleation arising from attrition fragments as a result of agitation and shear. Synthesis times evaluated were 3 h, 6 h, 24 h and 72 h for each combination with high water levels, and 1 h, 3 h, 6 h and 24 h for each low-water combination. All samples prepared in this study were replicated to confirm the reproducibility of the results. After recovery and drying of the product, the Na-form samples were characterised by selected methods as specified below, then detemplated and ion exchanged. Selected NH3 or H-form samples were then further characterised.
ZSM-5 product samples were characterised in replicate, as specified. First, all replicated ZSM-5 synthesis products were identified prior to detemplation and ion exchange by X-ray diffraction (XRD) for each time series to determine the purity of the mineral phase, the relative crystallinity and the relative time of formation of the fully crystalline zeolite phase. Powder X-ray diffractograms were obtained using a Philips X-ray diffractometer with Cu Kα radiation of wavelength 154 pm. The scan range was set to 5° < 2θ < 48°. The relative percentage crystallinity for all zeolites was obtained by comparing the summation of the intensities of the peaks, between 22° and 25° 2θ of a given sample, to the corresponding sum of a standard sample, which was chosen to be the most crystalline material. The relative percentage crystallinity of replicated samples was nearly identical in most cases.
The crystal morphology was determined by scanning electron microscopy (SEM). A Cambridge S200 was used to take micrographs of the catalyst samples obtained after 24-h synthesis times. The as-synthesised zeolites were mounted on aluminium stubs covered with a mixture of water-based glue and colloidal carbon, upon which a small dusting of each respective sample was deposited and coated with a thin film of Au/Pd. Particle sizing of ZSM-5 crystals was confirmed using a Malvern particle sizer.
The Si/Al ratio of the ZSM-5 crystals (24 h) was determined by atomic absorption spectroscopy (AAS). Al and Na, as well as K, contents of the un-detemplated, un-ion exchanged zeolites were analysed by using a Varian SpectrAA-30 spectrometer with a DS 15 data station. The following procedure was used: 100 mg zeolite was weighed into a Teflon-lined autoclave with a volume of 33 ml. Fifteen millilitres of concentrated hydrochloric acid (HCl, 37 wt%, Merck) was added carefully and afterwards the autoclave was sealed and placed in a hot-air oven at 150°C for 16 h. The samples were cooled to room temperature. An acidic supernatant and a SiO2 deposit were present in the Teflon liners. The contents of the Teflon liners were filtered; solids were collected by repeated washing and decanting of the supernatant and deposit onto an ashless filter paper, and the filtrate was collected in a Buchner flask for further analysis. The filtrate was transferred into a 50 ml volumetric flask and diluted to volume with H2O and then analysed for Al, Na and K. The filter paper and SiO2 deposit were ashed in a furnace at 1 000°C to constant weight using ceramic crucibles and the SiO2 content determined gravimetrically.
Thermogravimetric differential thermal analysis (TG/DTA) was used to determine the moisture content and the amount of incorporated template in the freshly synthesised ZSM-5 catalysts prior to detemplation and ion exchange. The TG/DTA analyses were obtained using a Stanton-Redcroft STA-780 thermal analyser. A sample mass of 50 mg of as-synthesised material (24 h) was heated at 10°C min–1 in nitrogen (30 ml min–1) to 600°C. This temperature was held for 1 h and then air (30 ml min–1) was passed over the sample at 600°C. The weight losses obtained from TGA below 200°C were ascribed to moisture loss and those at above 200°C to organic template material loss. The losses are presented as percentages of the total mass of synthesised catalyst.
After characterisation by XRD, SEM, TGA and AAS, the dry catalysts were converted from the Na-form into the NH3-form by detemplation and ion exchange. The ZSM-5 products were detemplated by placing samples in a muffle furnace to remove the organic template from the dried catalysts. The nitrogen flow into the chamber was set to 800 ml min–1 and the heat was ramped up to 500°C, at 10°C min–1, and held at that temperature for 8 h. Subsequently, the gas feed was switched to air at 500°C for a further 6 h. The detemplated Na-form ZSM-5 catalyst was ion exchanged to the NH3-form by placing the sample in a glass autoclave fitted with a reflux condenser. After the addition of an excess of 2 M ammonium nitrate solution, the solution was held at 90°C for 24 h and stirred magnetically. Finally, the catalyst was filtered, rinsed well with water and dried in an oven at 80°C for 24 h.
n-Hexane adsorption was used to determine sorption capacity of selected samples. Sorption of hydrocarbons was performed using a Stanton-Redcroft STA-780 thermal analyser fitted with a dosing system. This dosing system consisted of various saturators of different hydrocarbons, through which nitrogen was fed as the inert carrier gas. A sample mass of 50 mg of the detemplated, ion exchanged zeolite was heated at 10°C min–1 in air (30 ml min–1) to 500°C in order to calcine the zeolite. This temperature was held for 3 h in the case of n-hexane. Thereafter nitrogen (30 ml min–1), saturated with the sorbent at 0°C was passed over the sample at 80°C for 30 min, where the equilibrium state was reached.
Temperature-programmed desorption of ammonium (NH3-TPD) was performed in quartz sample cells containing 0.5 g of selected samples of ZSM-5 catalyst in the NH3-form. The catalyst was first calcined in flowing helium at 500°C for 24 h before being cooled in flowing helium to 150°C. NH3 was then adsorbed from a 4% NH3 in helium mixture for 60 min and the physisorbed NH3 was removed by purging in flowing helium for a further 24 h at 150°C. Temperature-programmed desorption (TPD) spectra were recorded by measuring the NH3 desorbed using a thermal conductivity detector (TCD) while increasing the temperature at 10°C min–1, from 150°C to 500°C, and maintaining the final temperature for 60 min. Baseline spectra (no NH3 adsorbed) were subtracted from all the ZSM-5 ammonia TPD spectra. NH3 mass balances, checked by titration of the exit steam, were always better than 95%.
Pore size distribution of samples obtained after 24-h synthesis time was determined by N2 porosimetry using the N2 BET technique. Sorption-desorption isotherms for nitrogen at 77 K were recorded using an automated porosimeter (Micromeritics Asap 2000). Surface areas were calculated from the BET equation. The total sorbed volumes, including adsorption in the micropores and mesopores and on the external surface, were calculated from the amount of nitrogen adsorbed at a relative pressure p/p0 before the onset of interparticle condensation. To obtain the size and the distribution of the mesopores the Barrett-Joyner-Halenda (BJH) method was used by calculating from the desorption branch of the isotherm.
Aluminium nuclear magnetic resonance spectroscopy was used to determine framework aluminium (FAl) and extra-framework aluminium (EFAl) of samples obtained after 24-h synthesis times. 27Al-NMR and 29Si-NMR were performed to determine the coordination of the aluminium and the local environment of the silicon using a Varian Unity 400 spectrometer operating at 104 MHz and 79.5 MHz, respectively. Spectra were acquired using a 7-mm ZrO2 rotor at ambient temperature. Typically, flip angles of ≈ 50° were used, with a 1-s pulse delay. Spinning speeds of 3-5 kHz and a field strength of B0 = 9.6 Tesla were applied. 27Al and 29Si chemical shifts are quoted relative to an external 1 000-ppm Al(NO3)3 solution or an aqueous solution of 3-(trimethylsilyl)tetradeuterosodium propionate (DDS), set to 0 ppm.
In order to characterise the activity of synthesised ZSM-5 samples obtained after a 24-h synthesis time in a high-water environment, the n-hexane cracking reaction was used. The samples were investigated in a fixed-bed plug flow reactor (PFR), whereby n-hexane was cracked into several products, via the monomolecular cracking mechanism, at a reaction temperature of 500°C and a low conversion was maintained. In the case of ZSM-5, no diffusion limitations and fairly little deactivation was expected. A fixed bed, consisting of the catalyst to be examined and sand, was placed in a stainless steel reactor. The mass ratio of catalyst:sand was 1:29. The height of the total catalyst bed was about 1.5 cm. The reactor had an inner diameter of 11 mm and a length of 300 mm. After loading the catalyst bed, the reactor was filled with pure sand from both flow directions and fitted with glass wool on the ends of the reactor tube. The temperature profile was measured by an axial movable thermocouple, placed in the centre of the tube. Samples for the hydrocarbon product flow analysis were taken by using an ampule system. Samples of 100 µl were injected with a gas syringe into a Varian 3700 gas chromatograph (GC) consisting of a HP1 capillary column, 50 m long with a diameter of 0.2 mm. The detector used in the GC was a flame ionising detector (FID). The temperature for the n-hexane cracking reaction was 500°C and the low partial pressure of n-hexane in the saturator was regulated to pn-hexane = 0.1 bar via regulating the temperature to –10°C. The ZSM-5 catalyst was calcined at 500°C for 3 h. Thereafter, the flow rate of the carrier gas nitrogen was set to 15 % (1.025 mmol min–1) and the cooling system of the n-hexane saturator was set to –10°C and stabilised for 30 min. Average activities were calculated after a 100-h time on-stream period.
Results and discussion
In a molar regime of 1–Al:100 Si:45 TPABr:22 MOH:y H2O: z anion with M=K or Na, y = 2 354 or 977 mol, z = 3 NO3– or 1.5 SO42–, which in all cases produced ZSM-5 as the only zeolitic phase (as determined by XRD), the following results were obtained by varying cation and anion source, as well as water content, systematically.
Time of formation
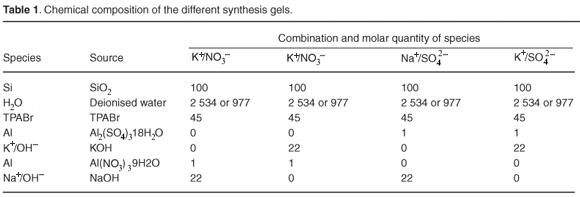
Distinctly different times of formation of fully crystalline ZSM-5 were obtained by varying cation and anion source as well as water content. These trends are shown in Fig. 1, which represents the relative percentage crystallinity obtained in the case of each consecutive time period for the series of ZSM-5 products derived from each different molar regime. The relative crystallinity was determined from XRD data through normalising the product crystallinity of all samples to the most crystalline product by the method described in the methods.
Irrespective of anion type and water content, Na+-containing combinations produced fully crystalline, pure ZSM-5 within 3 h. The K+/NO3– and K+/SO42– combinations were slower to form ZSM-5 in a high-water environment, taking about 6 h, after which they lost crystallinity.
When the K+/SO42– and K+/NO3– combinations were used in a low-water environment, highly-crystalline ZSM-5 only formed after 24 h. Crystal growth habit appeared to have been disrupted, with smaller, more intergrown particles resulting. Hence, the use of K+ as the cation retarded crystallisation, particularly in low-water environments. The greater viscosity of the low-water gels may also have contributed to hindered nutrient transport, thus contributing to the slower rates of formation observed for the K+ combinations. Hindered transport itself was not however the main cause of slow times of formation because no viscosity effect was observed in the low-water Na+ combinations. Low-water, high-viscosity Na+ combinations formed as quickly as the low-viscosity, high-water Na+ combinations. It is well documented that K+ is a structure-breaking cation, which this study confirms.
pH of molar regime
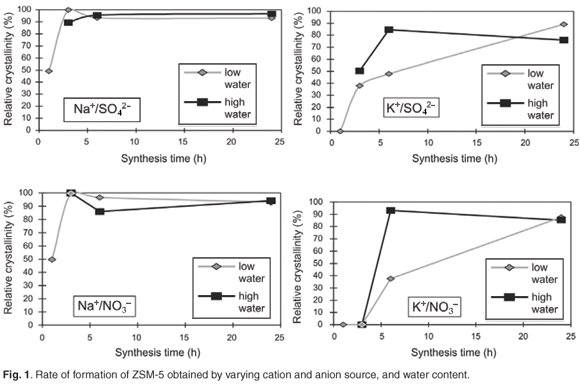
Figure 2 shows the replicated pH trends during hydrothermal synthesis that were obtained directly after each sample for each time series, was removed from the oven, rapidly cooled and opened.
Different and replicable pH trends were observed during hydrothermal synthesis for the different combinations obtained by varying cation and anion sources, as well as water content. In general, the presence of the nitrate anion created a more alkaline environment than the sulphate anion in the high-water environment. This is consistent with the fact that there was more nitrate than sulphate present and the results show that even small differences in anion concentration, due to the use of different sources of aluminium as mentioned previously, may change the synthesis environment considerably. The NO3– anion provided a consistently more alkaline environment than the SO42–. This effect was enhanced by use of the K+ cation. Lower pH trends were observed when using the Na+ cation than when using the K+ anion in high-water systems, possibly as a consequence of an increased buffering capacity of the system.
In the case of high water, the pH values increased rapidly within 3–6 h and decreased again to about pH 12.5 for combinations with K+/SO42– and K+/NO3–. This sharp increase to a high pH at the beginning of the hydrothermal synthesis period appeared to be detrimental to ZSM-5 formation as these combinations resulted in XRD amorphous material until a synthesis time of 24 h, by which time the pH had decreased. On the contrary, the pH for the combinations Na+/SO42– and Na+/NO3– barely increased within a 6-h synthesis time. These samples formed highly-crystalline ZSM-5 within 3 h.
The low-water environment resulted in a relatively lower pH synthesis environment. However, the cation type appeared to affect the pH in the first 5 h of synthesis for SO42– combinations. The NO3– anion stabilised both Na+ and K+ combinations at the same pH, although the relative difference in molar concentration of the anions was the same as in the high-water syntheses. The lower pH may also have resulted in slower depolymerisation and condensation of silica. Lower pH, i.e. fewer OH– ions, may change the rate of Si depolymerisation and hence the availability of Si species to participate in the subsequent condensation reactions, namely the formation of 3=3=Si–O–Si3=3= 'oxo'-bridges, and thus result in slower condensation.10–12
When the depolymerisation of the silica sol, and subsequent hydrolysis and condensation of the Si species onto the growing crystal was complete, the OH– species increased due to released OH– species for this process. With the formation of the zeolite products, the pH thus increased. The increasing pH trend was followed for Na+ combinations in the high-water regime and K+ combinations in the low-water regime.
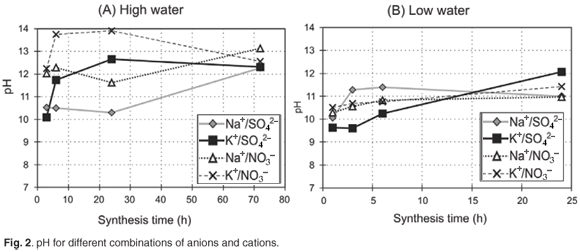
In order to obtain information on the transformation of the pre-synthesis gel into the product ZSM-5, the dry mass of the insoluble product obtained for each synthesis time after washing and drying was weighed carefully by using a milli balance. High product yield per mass of synthesis gel loaded was obtained for all combinations (Fig. 3). Product yield results obtained for each case were highly replicable.
In the case of the high-water synthesis, a general increase of the product yield can be observed over time. Except in the combination Na+/SO42–, all curves converge to a constant value of about 30–35 % yield after a 6-h synthesis time. The samples synthesised with K+ showed a yield of more than 30% after a 3-h synthesis time, whereas the samples synthesised with Na+ showed a range of somewhat lower values at a similar time. The nitrate anion in combination with Na+ appeared to enhance yield when compared to the combination containing the sulphate anion in the high water case, although the yield of product per mass of synthesis gel loaded is ultimately less. It is apparent, by comparison of the high yield values obtained for the potassium case with XRD and SEM results, that the high yield observed for K+ combinations is not associated with crystalline material and these products initially contained a significant amount of amorphous phase. The low-water combinations generally appeared to give high yields of 35–40% between 1 h and 6 h, which did not correlate with XRD crystallinity; yields decreased between 6 h and 24 h. The decrease of yield over time, observed in the case of K+/SO42– and K+/NO3–, probably is due to synthesis conditions favouring the dissolution of the amorphous solid material which formed initially in the case of the K+ combinations, compared to the competing process of condensation which forms the ZSM-5 product, which may be confirmed by the increasing trends in relative crystallinity observed by XRD. The cause of the decrease in yield, which is particularly noticeable in the case of potassium, was clarified by the SEM results which showed that dissolution of the crystals occurred and crystals underwent a change of morphology, indicating metastability, during synthesis.
Morphology, particle size, adsorption and pore size distribution
Table 2 and Fig. 4 summarise various characterisation data for the samples prepared in this study. These characterisation results were replicable. Scanning electron microscopy, Malvern particle sizing, and N2BET and n-hexane adsorption studies of the specified ZSM-5 products, prepared using the various combinations, showed that high-water Na+ combinations using either NO3– or SO42– resulted in the formation of spherical-stepped agglomerates of about 5 µm. However, the low-water environment changed the morphology of the ZSM-5 produced with these combinations, to intergrown, twinned crystals, of about 5 µm, which had a disrupted growth habit. All K+ combinations had a twinned morphology but anion type and water concentration resulted in different particle sizes. The K+/NO3– combination produced the largest crystals formed in this study, whereas the K+/SO42– combination had the same particle size as the Na+ combinations in high-water environments. Amorphous material was visible, by SEM and XRD, for low-water K+ combinations until 24 h. Nuclear magnetic resonance studies confirmed the presence of EFAl after 24 h in all samples prepared using K+ combinations, except for the low-water SO42– sample.
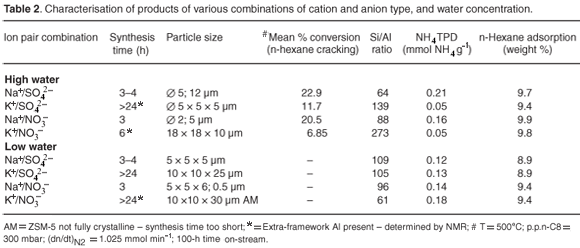
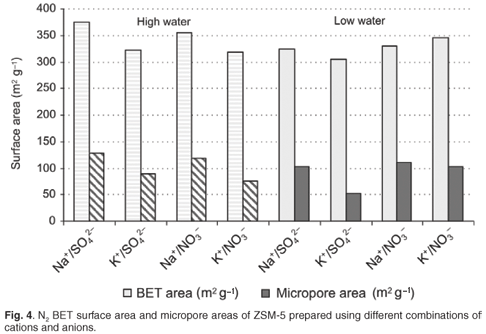
Although the Na+/NO3– and Na+/SO42– environment resulted in a crystal morphology of stepped agglomerates, that could be as a result of the inclusion of mother liquor during rapid crystal growth, potentially causing micropore blockage; this did not have a negative impact on the observed pore volumes or catalytic activity. Hexane adsorption indicated good access of reactant to the micropore area of ZSM-5 samples prepared rapidly in a high-water environment. Interestingly, the ZSM-5 prepared from NO3– combinations, particularly in a high-water environment, had the greatest hexane adsorption capacity. Overall trends observed for N2BET micropore and total surface areas of detemplated samples after 24-h synthesis times are shown in Fig. 4.
The most significant differences can be seen in the micropore surface areas. The samples prepared with K+ as the cation showed a significant lower micropore area in the high-water case. Whereas, for the K+/SO42– combination, an evident reduction in micropore and mesopore surface areas was observed in the low-water case. The nitrate anion counteracts the tendency to form a denser phase for K+ combinations in the low-water case. The sodium cation enhanced microporous surface areas of ZSM-5 products regardless of the anion combination. Low-water concentration played a role in reducing mesoporosity in combination with the K+ cation, as these combinations resulted in lower surface areas.
The pore size distribution from N2BET of different samples obtained after 24 h showed generally similar hysteresis behaviour with a hysteresis loop observed between a relative pressure of 0.5 p/p0 and 1 p/p0, and another hysteresis loop observed below a relative pressure of 0.2 p/p0 (Fig. 5).
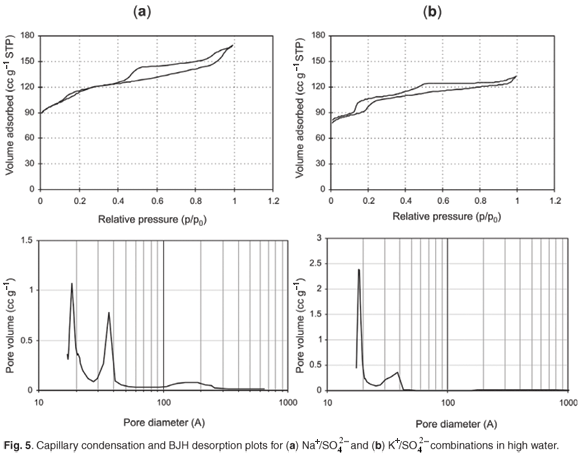
Hence a bimodal distribution of intracrystalline mesopores, with both smaller (~20 Å) and larger mesopores (~45 Å), was observed from BJH desorption data. In the case of the K+-containing combination, the formation of larger mesopores (~45 Å) was suppressed, possibly causing diffusional limitations, which may be why these samples exhibited a generally lower catalytic activity. For the samples prepared in this study, a high proportion of the available surface area reports to the non-shape selective mesoporous region. As enhanced mesoporosity is desirable in many catalytic reactions, this simple synthetic route is presented as a rapid and effective means in which to obtain a graded pore distribution.
Acidity, catalytic activity and Al environment
Table 2 shows the NH4 TPD data for ZSM-5 samples prepared using different combinations of cation and anion. The Na/SO42– combination prepared in a high-water environment displayed the highest acidity and catalytic activity, followed closely by the sample prepared with the Na+/NO3– high-water combination. Samples prepared using Na+ combined with NO3– or SO42–, in either high or low-water environments, exhibited no EFAl.
Extremely low acidities and reduced activities were obtained for ZSM-5 products prepared with the K+ cation, with either SO42– or NO3–, in a high-water environment, which may be related to the presence of EFAl as well as low acidity in these products. Elemental analysis of these products showed that Al remained largely unincorporated into the crystalline phase. Elemental analysis of post-synthesis supernatant liquors of these samples also showed that the reactant Al was wasted during synthesis. A low-water environment improved the incorporation of Al in products from K+-containing formulae. However, the presence of the sulphate anion inhibited the incorporation of Al in ZSM-5 products in low-water environments.
The acidities of the samples from NO3–-containing molar regimes which were prepared in a low-water environment were not compromised, but the acidity of samples prepared in the presence of SO42– were slightly reduced.
Extra-framework Al was present in the low-water K+/NO3– sample. Catalytic activity was not determined for these samples. The NMR and NH4TPD data, as well as the n-hexane cracking trends over 100-h time on stream periods, presented in Table 2, show that, although all molar regimes were loaded with a Si/Al ratio of 100, the various combinations of cations and anions resulted in differences in acidity and activity of the solid product. Differences in Si/Al ratio and the presence of EFAl resulted in changes in acidity, particularly in products obtained in a K+-containing high-water environment. This may be related to the different hydrothermal environments of each sample that resulted in disruption of the incorporation of tetrahedral Al into the crystalline phase.
Further to this, it is clear from the ratio of micropore area to total surface areas (including mesopores) presented in Fig. 4, that relatively more acid sites will be accessible on non-shape-selective mesoporous surface areas for samples of ZSM-5 prepared using Na+ in a high-water environment.
Conclusions
The characteristics of ZSM-5 were influenced significantly by merely changing the cation and anion type accompanying the hydroxide and alumina source during hydrothermal synthesis in a fixed molar regime. It is shown that under identical, replicable preparation and testing conditions the synthesis time, Al incorporation, product yield, acidity, morphology, pore size distribution and catalytic activity were affected. Rapid synthesis and mesoporosity of ZSM-5 were promoted by altering a fixed molar regime with respect to anion and cation type and water content only, particularly for Na+/NO3– combinations using standard hydrothermal synthesis conditions. An ion pair combination of Na+ and NO3– allowed the formation of pure ZSM-5 with enhanced mesoporosity and activity for n-hexane cracking in less than 3 h in a high-water environment. Use of a K+/NO3– ion pair combination in a high-water environment resulted in larger crystals, slower rates of formation, extra-framework Al and loss of acidity, as well as reduced n-hexane cracking activity. The sulphate anion and K+ inhibited the incorporation of Al. In this study, low water levels increased the time of formation of ZSM-5 and affected product quality, particularly in the presence of the sulphate anion. Hence, it is concluded that the molar regime for ZSM-5 should preferably contain the Na/SO42– or Na+/NO3– ion pair in a high-water environment for high Al incorporation. Optimum mesoporosity and acidity of ZSM-5 enhanced the activity for n-hexane cracking under these conditions.
We thank Matthias Knoll and the Catalysis Research Unit, Department of Chemical Engineering, University of Cape Town, South Africa.
1. Motuzas J., Julbe A., Noble R.D., Guizard C., Beresnevicius Z.J. and Cot D. (2005). Rapid synthesis of silicalite-1 seeds by microwave assisted hydrothermal treatment. Microporous Mesoporous Mater. 80(1–3), 73–83. [ Links ]
2. Inui T. (1989). Zeolite synthesis. In ACS Symposium Series, no. 398, eds M.L. Occelli and H.E. Robson, p. 479. American Chemical Society, Washington D.C. [ Links ]
3. Crea F., Aiello R., Nastro A. and Nagy J.B. (1991). Synthesis of ZSM-5 zeolite from very dense systems. Zeolites 11, 521–527. [ Links ]
4. Choudhary V.R. and Akolekar D.B. (1988). Crystallization of silicalite-factors affecting its structure, crystal size and morphology. Mater. Chem. Phys. 20(4–5), 299–308. [ Links ]
5. Nastro A. and Sand L.B. (1983). Growth of larger crystals of ZSM-5. Zeolites 3, 57–62. [ Links ]
6. Bhat Y.S., Das J., Rao K.V. and Halgeri A.B. (1996). Inactivation of external surface of ZSM-5: zeolite morphology, crystal size and catalytic activity. J. Catal. 159(2), 368–374. [ Links ]
7. Erdem A. and Sand L.B. (1979). Crystallization and metastable phase transformations of zeolite ZSM-5. J. Catal. 60(2), 241–256. [ Links ]
8. Petrik L.F., O'Connor C.T. and Schwarz S. (1995). The influence of various synthesis parameters on the morphology and crystal size of ZSM-5, and the relationship between morphology and crystal size and propene oligomerization. In Catalysis by Microporous Materials: Studies in Surface Science and Catalysis, vol. 94, eds H.K. Beyer, H.G. Karge, I. Kiricsi and J.B Nagy, pp. 517–524. Elsevier, Amsterdam. [ Links ]
9. Hardenberg T.A.J., Mertens L., Mesma P., Muller H.C. and Nicolaides C.P. (1992). A catalytic method for the quantitative evaluation of crystallinities of ZSM-5 zeolite preparations. Zeolites 12(6), 685–689. [ Links ]
10. Feijen E.J.P., Martens J. and Jacobs P. (1997). Hydrothermal zeolite synthesis. In Handbook of Heterogeneous Catalysis, vol. 1., eds G. Ertl, H. Knözinger and J. Weitkamp, pp. 311–323. Wiley-VCH, Weinheim. [ Links ]
11. Breck D.W. (1984). Zeolite Molecular Sieves: Structure, Chemistry, and Use. R.E. Krieger Publishing Company, Malabar. [ Links ]
12. Thompson R.W. (1998). Recent advances in the understanding of zeolite synthesis. In Molecular Sieves: Science and Technology, vol. 1, eds H.G. Karge and J. Weitkamp, pp. 1–33. Springer-Verlag, Berlin/Heidelberg. [ Links ]
Received 18 May. Accepted 26 May 2009.

