










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.105 n.1-2 Pretoria Jan./Feb. 2009
RESEARCH ARTICLES
Hereditary non-polyposis colorectal cancer is predicted to contribute towards colorectal cancer in young South African blacks
L. CronjéI, II; P.J. BeckerIII; A.C. PatersonI; M. RamsayII, *
IDivision of Anatomical Pathology, School of Pathology, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IIDivision of Human Genetics, School of Pathology, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IIIBiostatistics Unit, Medical Research Council of South Africa, Pretoria, South Africa
ABSTRACT
A disproportionately large number of young (<50 years) black patients present with colorectal cancer (CRC) in South Africa. Although a phenomenon previously described elsewhere in Africa, its specific molecular basis, whether sporadic or hereditary, has not been established. Molecular analysis of these tumours could link them to the features known to be associated with specific types of hereditary colorectal cancer, specifically through examination of levels of microsatellite instability, promoter methylation and the presence or absence of KRAS and BRAF mutations. The molecular features of cancer tissue samples from 44 CRC cases of black and white patients in South Africa were accordingly retrospectively analysed without knowledge of family history. Compared with samples from older blacks (>50 years), those from young black patients presented more often with a low methylation phenotype (CIMP-L) and high levels of microsatellite instability (MSI-H). Furthermore, as determined by real-time PCR using probe technology, the tissues from 35% of young blacks showed mutations within exon 1 of the KRAS gene. The BRAF-V600E mutation was only evident in the case of a single young black patient. Based on these results it seems likely that a proportion of CRC cases in young black patients from South Africa develop through the accumulation of mutations resulting in a mismatch repair deficiency linked to MSI-H and, possibly, germline mutations in the mismatch repair genes. The features in these patients are consistent with a diagnosis of the Hereditary Non-Polyposis Colorectal Cancer (HNPCC) syndrome. This finding has important implications for patient management and suggests that family members may be at high risk for CRC.
Key words: colorectal neoplasms, hereditary cancer syndromes
Introduction
Colorectal carcinogenesis involves the stepwise accumulation of mutations and/or epigenetic alterations, leading to the transformation of normal colonic epithelia. This process may develop and progress over a period of 10 to 15 years. Comprehensive studies have examined both morphological and molecular changes associated with the initiation and progression of colorectal cancer. A wealth of knowledge has thus far been accumulated and has led to the detailed classification of four distinct molecular pathways.
The classical pathway, somewhat involved in all of the pathways described, involves the somatic mutational inactivation of the adenomatous polyposis coli (APC) gene in colorectal epithelial cells.1 This leads to a cascade of events including, amongst others, degradation of β-catenin binding sites and interference with E-cadherin homeostasis during tumour initiation, and ultimately to p53 gene mutations during tumour progression.2 The familial form of this pathway is the autosomal dominantly inherited predisposition to familial adenomatous polyposis (FAP) that is initiated by germline mutations in the APC gene, and characterised by the presence of adenomatous polyps which develop into colorectal cancer if left untreated.3
The next pathway involves the accumulation of mutations due to a mismatch repair (MMR) deficiency, resulting in microsatellite instability (MSI) in the coding regions of genes implicated in tumour progression.4 This may lead to differential levels of MSI, differentiated by the number of tested loci displaying instability.5 Tumours with high levels of instability (MSI-H) may develop on a hereditary basis, involving the MMR genes hMSH2, hMLH1, hMSH6, hPMS2 and hPMS1, and thus predispose to hereditary non-polyposis colorectal cancer (HNPCC).6
The so-called 'serrated' pathway involves the silencing of MMR genes through promoter hypermethylation.7 This pathway is initiated in 'serrated' neoplasia through the inhibition of apoptosis, followed by the disruption of DNA repair mechanisms through epigenetic silencing.7
There is a worrisome trend in South Africa for a disproportionately large number of young black patients to present with CRC.8,9 The specific morphological features of the tumours concerned do not seem to indicate the involvement of diet or lifestyle-related factors, and investigations into their pathogenesis may prove to be useful to establish molecular markers for early detection and treatment. This study aimed at investigating CRC in black patients to identify the possible molecular pathways involved, to aid in the establishment of molecular biomarkers. Features such as overall methylation phenotype (CIMP), microsatellite instability (MSI) and BRAF and KRAS gene mutation status were investigated. Results were obtained through methylation-specific polymerase chain reaction (MSP-PCR), microsatellite analysis and real-time PCR.
Materials and methods
Patient selection
Paraffin-embedded tissue samples were collected retrospectively, and included specimens originally diagnosed in the Division of Anatomical Pathology as adenocarcinoma of the colon and/or rectum. Cases were limited to a randomly selected subset of the more recent cases (1999–2003) based on tissue availability, and were stratified according to age at diagnosis (<50 years or >50 years) and ethnicity (black or white), with 44 patients investigated in detail: 25 blacks (<50 years); three whites (<50 years); eight blacks (>50 years); and eight whites (>50 years). The study was approved by the Ethics Committee of the University of the Witwatersrand for retrospective work on archival material (clearance number 9/11/88).
DNA extraction
In each case, two blocks were identified that contained exclusively either non-tumourous normal or tumourous material. The non-tumourous normal material was collected during the resection at a position 10 cm removed from the macroscopically identified tumour. Serial sections of 10 µm of each identified tissue block were collected in separate tubes. DNA extraction was performed through the phenol-chloroform method. Briefly, cut sections were de-waxed using xylene before the addition of lysis buffer (50 mM Tris base (Saarchem, Wadeville, South Africa), 5 mM EDTA (Saarchem, Wadeville, South Africa), Tween20 (Sigma Chemical Company, St Louis, MO), 10% proteinase K (Roche Diagnostics, Penzburg, Germany); pH 8). Following an overnight incubation at 56°C the proteinase K was inactivated at 80°C for 10 min. Phenol-chloroform (1:1) was subsequently added, vortexed briefly and centrifuged (16.1 × g, 4 min) (Eppendorf, model 5415R) before adding the top organic layer to a separate tube containing chloroform (Sigma Chemical Company, St Louis, MO). This was briefly vortexed and centrifuged (16.1 × g for 4 min), before adding the top layer to a mixture of iso-propanol (Saarchem, Wadeville, South Africa) and 3 M sodium acetate (Saarchem, Wadeville, South Africa). This was carefully inverted and centrifuged (16.1 × g, 30 min at 4°C), the DNA-pellet washed with 70% ethanol (Saarchem, Wadeville, South Africa), and dried at 65°C for 2 min. This was then dissolved in TE buffer, pH 8.4 (0.04 mM Tris base, 0.05 M EDTA pH 8), incubated at 65°C for 10 min, and stored at 4°C until further use. Yield was determined using the NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). The integrity of the isolated DNA was routinely evaluated through the amplification of the β-globin gene using the primer pair PC04/GH20 (Table 1).

Methylation-specific PCR
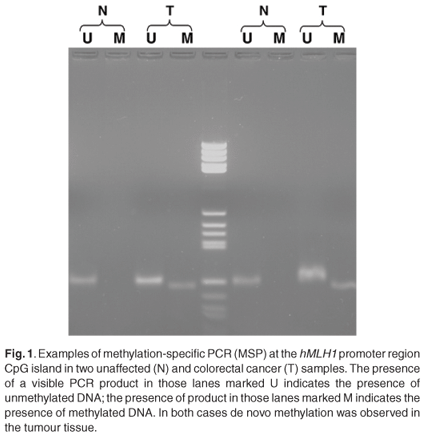
The methylation status of MINT1, MINT2, MINT31, hMLH1 and MGMT was determined by bisulphite treatment of DNA. The methylation status of both non-tumour and tumour tissue of each case was determined separately. These loci were chosen based on published data that showed they offered a means of discriminating the CpG island methylator phenotype (CIMP), and that the MINT loci were unmethylated in normal tissues.10 Bisulphite modification was performed using a commercially available kit (CpGenomeTM DNA modification kit, Chemicon Int, Temecula, CA) according to the manufacturer's instructions. Methylation-specific PCR (MSP) was done using primers specific for either the methylated (M) or modified unmethylated (U) DNA described in Table 1. Appropriate positive and no-template DNA reaction controls were included in each PCR experiment. The products were visualised on a 3% agarose gel (Saekem LE, Cambrex BioScience, Rockland, ME; MS-4 agarose for MGMT reaction products, Whitehead Scientific, Cape Town, South Africa) (80 V, 80 min) stained with ethidium bromide. The loci were classified as unmethylated if the intensity of the methylated band was visually less than that of the unmethylated band, or as methylated if the intensity of the methylated band was visually more than that of the unmethylated band. CIMP status was determined as CIMP-negative if none of the evaluated loci were methylated, CIMP-low if one locus was methylated, and CIMP-high if two or more loci were methylated.
Microsatellite instability (MSI) testing
The loci recommended by the American Joint Commission on Cancer and the International Collaborative Group on HNPCC for microsatellite instability testing include BAT25, BAT26, D5S346 (APC), D17S250 (Mfd15CA) and D2S123.5 The MSI status of these loci was detected using a multiplex PCR system developed by Roche Diagnostics (HNPCC Microsatellite Instability Test, Catalogue number 2 041 901, Roche, Mannheim, Germany). One hundred nanograms of DNA were used to amplify the MSI loci in a single multiplex reaction using the provided primer pairs labelled with different fluorophores (6-FAM, TET or HEX). Analysis of PCR products was performed on the ABI PRISM automated sequencer model 377 (Applied Biosystems, Foster City, CA) according to manufacturer instructions. MSI was defined by the presence of novel peaks, following the PCR amplification of tumour DNA, which was not present in non-tumour DNA. A tumour was classified as high-MSI (MSI-H) if at least 40% (2/5) of the examined loci showed unequivocal instabilities.5 Microsatellite-stable (MSS) tumours were those where no microsatellite instability was found, while tumours with only one marker showing instability were declared microsatellite instability low (MSI-L).
Real-time PCR analysis for KRAS and BRAF gene mutation status
Detection of mutations in codon 12 and 13 of exon 1 of the KRAS gene (GGT to GAT transition), and in exon 15 of the BRAF gene (T1796A or V600E mutation) was performed using real-time PCR and melting curve analysis. Positive controls included DNA from a papillary thyroid carcinoma for the BRAF V600E mutation, while an oesophageal carcinoma served as control for KRAS mutation.11,12 The primers and fluorescently-labelled probes (MetaBion International, Martinsried, Germany) were designed using the LightCycler Probe Design Software (Version 2.0, Roche, Mannheim, Germany) (Table 1). PCR was performed in 20 µl volumes consisting of 3 mM MgCl2, 0.5 µM of each primer, 0.2 µM of each probe and 2 µl of LightCycler FastStart DNA master HybProbe (Roche, Mannheim, Germany). Following amplification (Software Version LCS4 4.0.0.23, Roche, Mannheim, Germany) the reaction mixtures were denatured and slowly heated to 80°C at a ramping rate of 0.1°C s–1. Melting peaks were subsequently constructed by plotting the negative derivative of the fluorescence with respect to temperature (-dF/dT). Results were randomly verified through DNA sequencing, using the Applied Biosystems 3130 Genetic analyser utilising capillary-based technology.
Statistical analysis
Statistical comparisons between the ethnic groups were completed using the two-sided Fisher's exact test. Patients were analysed based on ethnicity (Black vs White) and age (<50 years vs >50 years). All statistical analyses were completed using Stata Intercooled 7.0 (Stata, College Station, TX, USA). The differences were considered statistically significant when P < 0.05.
Results
Table 2 summarises the results obtained and data available on the sample population.

Methylation studies
The overall methylation status was determined by examining five distinct loci. These included the methylated in tumour (MINT) markers 1, 2 and 31, as well as the promoter regions of the hMLH1 and MGMT genes. Although not statistically significant, a high level of the CpG island methylator phenotype (CIMP-H) was more common in tumours located on the right in the young black patients [6/13 (P = 0.46) compared to 2/11 (P = 0.18) for left-sided tumours]. The other groups were too small to predict a trend. The majority of young blacks presented with low methylation status (CIMP-L) [16/25; P = 0.64) (Fig. 1)]. Methylation of the MGMT promoter region was often observed in young blacks (19/36; P = 0.41). Methylation of the hMLH1 promoter region was found in only 28% of young blacks (7/25), however. The positive predictive values of loss of protein expression of the MGMT and hMLH1 were 0.79 and 0.84, respectively (Fig. 1).
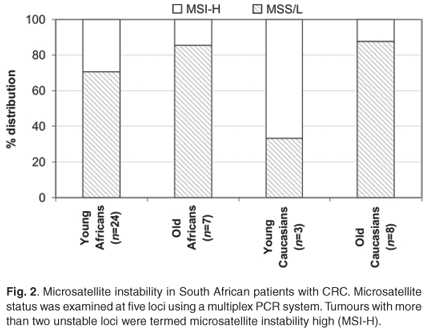
Microsatellite instability
The distinction between the mutator and methylator pathways is characterised by the microsatellite instability (MSI) status of key markers. MSI was analysed at the five recommended loci and included Bat25, Bat26, D5S346 (APC), D17S250 (Mfd15CA) and D2S123. Overall, in agreement with features of the mutator pathway, MSI-H was more often observed in young patients (7/25 black and 2/3 white) in comparison with patients older than 50 years of age at diagnosis (1/7 blacks and 1/8 whites). There appeared to be a higher level of MSI-H in the young patients, but this was not statistically significant (Fig. 2). In young black patients, these tumours were more frequently observed proximal to the splenic flexure (Table 2).8
BRAF and KRAS gene mutation status
Only one of the young black patients (1/25) presented with the V600E mutation within the BRAF gene, while 32% (8/25) showed mutations within the KRAS gene (Table 3). Interestingly, none of the young white patients (0/3) presented with either of the BRAF or KRAS gene mutations that were examined. Amongst older blacks and whites, however, 25% each (2/8 in each group) showed KRAS gene mutations, and no BRAF gene mutations were detected.

Combined morphological and molecular data
The data presented in Table 2 emanate from the present study and a previous study by Cronje et al.8 The following features were present at significant frequencies in patients with HNPCC: tumour site 'R'; tumour grade 'H', mucin expression 'H', MSI status 'H', loss of MMR protein (hMLH1, hMSH2 and/or hMSH6) 'Y', CIMP status 'L', BRAF mutation negative and KRAS mutation positive. When examined for each of these features, seven of the young black CRC patients (patient numbers 1, 2, 5, 6, 12, 13 and 15) and one young white CRC patient (patient number 26) exhibited over half of the features characteristic of HNPCC. None of the older patients exhibited more than half of these features.
Discussion
This study investigated the specific molecular features associated with colorectal cancer (CRC) in a group of South African patients. A previous comprehensive study on the morphological features associated with CRC in these patients had revealed that young black patients appeared to present with morphological features associated with HNPCC in a significant proportion of cases.8 These included proximally located (right-sided), high-grade tumours with a mucinous appearance, as reported for HNPCC tumours.13 The molecular features associated with this disease include mutations within the mismatch repair genes, mainly hMLH1 and hMSH2 that subsequently result in high levels of microsatellite instability (MSI-H). Diagnosis of HNPCC mainly relies on the Amsterdam and Bethesda criteria of which several revised editions are available.14 As a purely retrospective study, a lack of available family history complicated the diagnosis of HNPCC based on the above-mentioned criteria. This is often the case in academic, provincial and rural hospitals in developing countries such as South Africa where language barriers, inadequate understanding of the causes of death, or a lack of diagnosis are often encountered as limiting factors. We therefore postulated that through the use of molecular biology the diagnosis could be made directly in association with the morphological features identified.
HNPCC is an autosomal dominant cancer-susceptibility syndrome that accounts for approximately 1% to 6% of all CRC cases diagnosed.15 It is recognised at an early age (~45 years), in multiple individuals within a family and arises from adenomas with an advanced transformation rate, which is due to mutation and the subsequent inactivation of DNA mismatch repair genes, especially hMLH1 and hMSH2.15 Loss of immunohistochemical nuclear expression of these gene products often indicate the germline-inactivated gene, and it was previously reported that young blacks frequently showed loss of expression of the hMLH1 (23%; 29/128) (P = 0.121) and hMSH2 (12%; 16/129) (P = 0.013) proteins.8 In the 25 young black CRC patients of this study, nine (36%) showed such a loss of expression (Table 2). A hallmark feature used in the molecular diagnosis of HNPCC is the high level of microsatellite instability.16 Twenty-nine per cent of the young black patients in the current study presented with MSI-H in comparison with only 14% of older black patients (Fig. 1). It is of interest that two of the three young whites presented with the same molecular phenotype but did not show the morphological features associated with an HNPCC diagnosis. This is partially explained through the serrated neoplasia pathway that involves high levels of MSI and frequently methylated DNA regions.
The concept of serrated neoplasia is well established and involves the methylation and subsequent silencing of CpG islands often found in gene promoter sequences.7 Genes silenced in this way include amongst others the hMLH1 and DNA repair gene O-6-methylguanine DNA methyltransferase (MGMT), that is associated with high or low levels of MSI respectively.17 Only one of the 25 young black patients showed methylation within the hMLH1 gene in conjunction with the MSI-H phenotype.
KRAS gene mutations in codon 12 and 13 of exon 1 were frequently observed in young blacks (Table 3). A mutually exclusive relationship has been reported for the KRAS and BRAF-V600E mutations, which was confirmed by the current series where only one out of 25 cases in young black patients presented with the BRAF–V600E mutation (Table 3).18 Deng et al. recently showed that tumours from HNPCC patients do not harbor the BRAF–V600E mutation.19 This finding was subsequently confirmed by Domingo et al., who suggested BRAF screening as a strategy for simplifying HNPCC genetic testing since the presence of the BRAF mutation would suggest an alternative explanation.20 During the same time, Oliveira et al. showed that KRAS is mutated in 40% (63/158) of HNPCC families that contain germline mutations in one of the mismatch repair genes.21
BRAF and KRAS gene mutation status can conveniently be used to distinguish between hereditary CRC in the form of HNPCC and sporadic MSI-H CRC. The main distinguishing factor between these two possibilities is the presence of promoter methylation within the hMLH1 gene found in sporadic CRC. Several groups have shown a relatively low frequency of KRAS mutations in sporadic CRC in a background setting of hMLH1 promoter methylation when compared to HNPCC.22 The methylation status in young black patients reported here was predominantly low, with infrequent methylation of the hMLH1 and MGMT promoter regions. Therefore the frequent mutation of the KRAS gene observed in young patients, together with high levels of microsatellite instability, provides support for the hypothesis that these patients may present with a hereditary form of CRC, most likely HNPCC.
The diagnosis of HNPCC is predominantly based on fulfilment of the revised forms of the Amsterdam and Bethesda criteria, pertaining primarily to a family history of colorectal cancer.13 As stated earlier, it is often problematic to obtain family history from affected individuals in developing countries due to factors such as language barriers, fears of isolation and being prejudiced against and overall patient reluctance to follow-up after initial diagnosis. The need therefore exists for diagnostic criteria that rely on morphological and molecular features for accurate diagnosis. Studies into the morphological features of colorectal cancer in young black patients have revealed features in keeping with the sensitive Bethesda criteria for a diagnosis of HNPCC with the exception of family history.8 These include, amongst others, poorly differentiated carcinomas arising in the proximal colon.23 Taken together with the results reported here, which described high levels of MSI with limited methylation against a background of increased KRAS mutation, it seems evident that a subset of these young blacks with colorectal cancer presented with features associated with HNPCC. An analysis of the criteria presented in Table 2 shows that seven of the 25 (28%) presented with over half the characteristic features for HNPCC, whereas none in the >50 years age group (black and white) presented with more than four of the eight characteristics. Even though the present study included a modest number of young black patients, the clinical relevance of this study cannot be underestimated. Clinicians are therefore urged to obtain a comprehensive family history, and to test for HNPCC through germ-line mutations in the hMLH1 and hMSH2 genes, if the Amsterdam criteria are not met.
The authors wish to acknowledge the H.E. Griffin Trust and the Medical Research Council of South Africa for providing funding for this study.
1. Hamilton S.R., Vogelstein B., Kudo S., Riboli E., Nakamura S. and Hainout P. (2000). Tumours of the colon and rectum. In Pathology and Genetics of Tumours of the Digestive System, eds S.R. Hamilton and L.A. Aaltonen, pp. 103–129. IARC Press, Lyon. [ Links ]
2. Bright-Thomas R.M. and Hargest R. (2003). APC, β-catenin and hTCF-4; an unholy trinity in the genesis of colorectal cancer. Eur. J. Surg. Oncol. 29, 107–117. [ Links ]
3. Fearnhead N.S., Britton M.P. and Bodmer W.F. (2001). The ABC of APC. Hum. Mol. Genet. 10, 721–733. [ Links ]
4. Raut C.P., Pawlik T.M. and Rodriquez-Bigas M. (2004). Clinicopathologic features in colorectal cancer patients with microsatellite instability. Mutat. Res. 568, 275–282. [ Links ]
5. Boland C.R., Thibodeau S.N., Hamilton S.R., Sidransky D., Eshleman J.R., Burt R.W., Meltzer S.J., Rodriques-Bigas M.A., Fodde R., Ranzani G.N. and Srivastava S. (1998). A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 58, 5248–5257. [ Links ]
6. Peltomäki P. and de la Chapelle A. (1997). Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. Gastroenterol. 113, 1146–1158. [ Links ]
7. Jass J.R., Walsh M.D., Barker M., Simms L.A., Young J. and Legget B.A. (2002). Distinction between familial and sporadic forms of colorectal cancer showing DNA microsatellite instability. Eur. J. Cancer 38, 858–866. [ Links ]
8. Cronjé L., Paterson A.C. and Becker P.J. (2009). Colorectal cancer in South Africa: a heritable cause suspected in many young black patients. S. Afr. Med. J. 99, 103–106. [ Links ]
9. Boytechev H., Markovic S. and Oettle G.J. (1999). The characteristics of large bowel cancer in the low-risk black population of the Witwatersrand. J. R. Coll. Surg. Edinb. 44, 366–370. [ Links ]
10. Toyota M., Ahuja N., Ohe-Toyota M., Herman J.G., Baylin S.B. and Issa J.P. (1999). CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 96, 8681–8686. [ Links ]
11. Xing M. (2005). BRAF mutation in thyroid cancer. Endocr. Relat. Cancer. 12, 245–262. [ Links ]
12. Lord R.V.N., O'Grady R., Sheehan C., Field A.F. and Ward R.L. (2000). K-ras codon 12 mutations in Barrett's oesophagus and adenocarcinomas of the oesophagus oesophagogastric junction. J. Gastroenterol. Hepatol. 15, 730–736. [ Links ]
13. Umar A., Boland C.R., Terdiman J.P., Syngal S., de la Chapelle A., Rüschoff J., Fishel R., Lindor N.M., Burgart L.J., Hamelin R., Halimton S.R., Hiatt R.A., Jass J., Lindblom A., Lynch H.T., Peltomaki P., Ramsey S.D., Rodriquez-Bigas M.A., Vasen H.F., Hawk E.T., Barret J.C., Freedman A.N. and Srivastava S. (2004). Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer. Inst. 96, 261–268. [ Links ]
14. Grady W.M. (2003). Genetic testing for high-risk colon cancer patients. Gastroenterol. 124, 1574–1594. [ Links ]
15. Lynch H.T. and de la Chapelle A. (2003). Hereditary colorectal cancer. N. Engl. J. Med. 348, 919–932. [ Links ]
16. Rowley P.T. (2005). Inherited susceptibility to colorectal cancer. Annu. Rev. Med. 56, 554. [ Links ]
17. Chan A.O., Issa J.P.H., Morris J.S., Hamilton S.R. and Rashid A. (2002). Concordant CpG island methylation in hyperplastic polyposis. Am. J. Pathol. 160, 529–536. [ Links ]
18. Kim I.J., Kang H.C., Jang S.G., Kim K., Ahn S.A., Yoon H.J., Yoon S.N. and Park J.G. (2006). Oligonucleotide microarray analysis of distinct gene expression patterns in colorectal cancer tissues harboring BRAF and K-ras mutations. Carcinogenesis 27, 392–404. [ Links ]
19. Deng G., Chen A., Hong J., Chae H.S. and Kim Y.S. (1999). Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer. Res. 59, 2029–2033. [ Links ]
20. Domingo E., Laiho P., Ollikainen M., Pinto M., Wang L., French A.J., Westra J., Frebourg T., Espin E., Armengol M., Hamelin R., Yamamoto H., Hofstra R.M., Seruca R., Lindblom A., Peltomäki P., Thibodeau S.N., Aaltonen L.A. and Schwartz S. (2004). BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J. Med. Genet. 41, 664–668. [ Links ]
21. Oliveira C., Westra J.L., Arango D., Ollikainen M., Domingo E., Ferreira A., Velho S., Niessen R., Lagerstedt K., Alhopuro P., Laiho P., Veiga I., Teixeira M.R., Ligtenberg M., Kleibeuker J.H., Sijmons R.H., Plukker J.T., Imai K., Lage P., Hamelin R., Albuquerque C., Schwartz S., Lindblom A., Peltomaki P., Yamamoto H., Aaltonen L.A., Seruca R. and Hofstra R.M. (2004). Distinct patterns of KRAS mutations in colorectal carcinomas according to germline mismatch repair defects and hMLH1 methylation status. Hum. Mol. Genet. 13, 2303–2311. [ Links ]
22. Jass J.R., Biden K.G., Cummings M.C., Simms L.A., Walsh M., Schoch E., Meltzer S.J., Wright C., Searle J., Young J. and Legget B.A. (1999). Characterisation of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways. J. Clin. Pathol. 52, 455–460. [ Links ]
23. Giardiello F.M., Brensinger J.D. and Petersen G.M. (2001). AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterol. 121, 198–213. [ Links ]
Received 6 May 2008. Accepted 27 February 2009.
* Author for correspondence. E-mail: michele.ramsay@nhls.ac.za

