










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.104 n.1-2 Pretoria Jan./Feb. 2008
RESEARCH LETTERS
Effect of macromolecular crowding on the stability of monomeric glutaredoxin 2 and dimeric glutathione transferase A1-1
Diane C. Kuhnert; Samantha Gildenhuys; Heini W. Dirr
Protein Structure-Function Research Unit, School of Molecular and Cell Biology, University of the Witwatersrand, Private Bag 3, WITS 2050, South Africa
ABSTRACT
The effect of macromolecular crowding on the structure and stability of monomeric glutaredoxin 2 (Grx2) and its homodimeric structural homologue human glutathione transferase A1-1 (hGST A1-1) was investigated using dextran 70 as crowding agent. Far-UV circular dichroism and fluorescence spectroscopic data indicated that repulsive steric interactions between the proteins and dextran (50-300 mg/ml) had little effect on the global structures of the native proteins. Urea-induced unfolding of both proteins was reversible (recoveries of >80%) at low dextran concentrations (<100 mg/ml) but resulted in significant losses in refolding recoveries at higher levels of dextran, due to aggregation. The two-state global unfolding processes of Grx2 and hGST A1-1, as well as their m-values (unfolding cooperativity parameter), were unaffected by 100 mg/ml dextran, demonstrating the absence of specific intermolecular interactions between protein and crowder. Dextran at 100 mg/ml enhanced the stability of Grx2 and hGST A1-1 by 1.1 kcal/mol and 2.2 kcal/mol, respectively. Compaction of the unfolded states of both proteins is indicated by an increase in alpha-helical content and in the decreased solvent exposure of their tryptophan residues. The dextran-induced formation of compact states of urea-denatured Grx2 and hGST A1-1 is ascribed to steric excluded volume effects, which induce an entropic destabilization of expanded unfolded states, thereby shifting the equilibrium between native and unfolded states towards the native state. Quantitatively, however, the extent of stabilization of Grx2 is lower than that predicted by the equivalent hard particle model for the excluded volume effect of dextran on protein stability.
Introduction
The majority of biochemical transactions performed in organisms are carried out by proteins, the functions of which are dependent upon their three-dimensional structures. The shape assumed by a protein molecule, and its stability, in turn, is determined by numerous environmental factors, such as molecular crowding. Although the environment within cells and biological fluids is highly crowded by macromolecules,1 and because the excluded volume effect is a fundamental characteristic of crowded solutions,2,3 most studies on protein stability and function are performed with dilute protein solutions in the absence of other macromolecules. In order to understand the behaviour of globular proteins in vivo better, it is important to investigate the influence of a crowded environment on their stabilities and functions.
It was recently shown that the thermodynamic stabilities of two monomeric proteins, ( repressor and cellular retinoic acid-binding protein I, measured in vivo are very similar to those measured under in vitro dilute conditions.4,5 The larger m-values (i.e. δΔG/δ[urea]) observed in vivo for both proteins, however, suggests mechanistic differences between their in vitro and in vivo unfolding processes,5 and that they may be significantly influenced by attractive interactions between proteins and cellular components such as molecular chaperones.6,7
Various theories have been developed regarding the effects of excluded volume by macromolecular crowding on the stability of globular proteins, although limited in the context of the heterogeneity of biological systems, in which complex types of intermolecular interactions occur. These theories predict the stabilizing effects caused by inert crowders to be either small8,9 or large.3,10 The former model predicts that macromolecular crowding adversely affects both folded and unfolded states, whereas the latter models predict that the compact native state becomes significantly stabilized relative to an ensemble of more expanded unfolded states. Further, in vitro experiments have also demonstrated small to large stabilizing effects of excluded volume on protein stability.11-13 While protein denaturation studies have provided direct experimental evidence that soluble crowding agents induce a destabilization and compaction of expanded unfolded states, thereby shifting the equilibrium between native and unfolded states towards the native state,13,14 theory predicts semi-quantitatively the observed crowder-induced stabilization of only a few proteins.3,13,14 Recent theoretical analyses have shown macromolecular crowding and confinement to affect the stability of proteins to a similar extent.3,10 Experimental studies demonstrate that the confinement of proteins within the nanopores of silica or polyacrylamide gels does not enhance the stability of proteins substantially15 and that confinement, like that observed in vivo, can alter the mechanism of protein unfolding/refolding.16,17 The latter is suggested by the significant differences between the dependencies of the free energy of unfolding upon denaturant for confined and unconfined proteins. The large (25-32°C) increase in the Tm of α-lactalbumin confined in silica gel should not necessarily be interpreted as a substantial increase in the ΔGN-D of the entrapped protein, as its thermal unfolding transition displays a much reduced slope compared to that of the protein in solution, since such changes can result from destabilization of a protein.18
Considering the range observed of the effects of crowding on protein stability, and that very few experimental data on reversible protein-folding models are available to test excluded volume theories, more experimental data are required to understand better the effects of macromolecular crowding on protein stability and for the development and refinement of predictive models. In this study, we have used two homologous proteins (<10% sequence identity), Escherichia coli glutaredoxin 2 (Grx2) and human glutathione transferase A1-1 (hGST A1-1), to investigate the effects of macromolecular crowding on protein stability. While hGST A1-1 is homodimeric (50 kDa; 222 residues per subunit),19 Grx2 is monomeric (215 residues) and structurally resembles the subunit of canonical GSTs.20,21 In the absence of volume exclusion effects, both proteins unfold reversibly in the presence of urea. hGST A1-1 unfolds via a three-state equilibrium process, N2 ↔ N2* ↔ 2U, where N2 represents native dimer, N2* is an inactive native-like dimer with an unfolded helix 9, and U represents unfolded monomer.22 The first unfolding event represents the local unfolding of helix 9, which does not impact on the overall stability of the protein.23 Equilibrium unfolding of Grx2 is also a two-state process, N ↔ U, where N represents the native monomer and U is the unfolded monomer (Gildenhuys, Wallace and Dirr, unpublished work). The effects of macromolecular crowding by dextran on the native and unfolded states of both proteins were examined by tryptophan fluorescence and far-UV circular dichroism (CD) spectroscopy. The stability of each protein in the absence and presence of dextran was determined by urea-induced unfolding under highly reversible conditions, and the extent of stabilization by volume excluded effects compared to that predicted by theory. Given that the stability of helix 9 at the active site of hGST A1-1 contributes significantly towards catalytic function, the effect of macromolecular crowding on catalytic function was studied and compared with that of a class Mu enzyme, rGST M1-1, for which there is no corresponding helix at its active site.
Materials
See Appendix.
Results and discussion
Dextran 70, which represents a model of rigid rods,3 was chosen as a crowding agent due to its uncharged and inert nature and because it influences the behaviour of proteins essentially via nonspecific, repulsion interactions (that is, excluded volume effects).29,30 Further, a statistical-thermodynamic model for the excluded volume effect on protein stability has been developed for dextran,3,13 which allows a comparison of experimental and theoretical data.
Stabilization of Grx2 and hGST A1-1 by volume exclusion
The spectroscopic data shown in Fig. 1 indicate that dextran 70 at 50-300 mg/ml does not affect the native state of either Grx2 or hGST A1-1. This is demonstrated by the independence of the secondary structure (ellipticity at 222 nm) and the tertiary environments of tryptophan residues (fluorescence emission maximum wavelength) on the concentration of dextran. Since repulsive steric interactions between protein and crowder contribute negligibly towards the total energy of the proteins' native states, the effect of crowding on the thermodynamic stability of Grx2 and hGST A1-1 was investigated to assess the hypothesis that the unfolded state of a protein can adopt a more stable compact structure under conditions of macromolecular crowding.
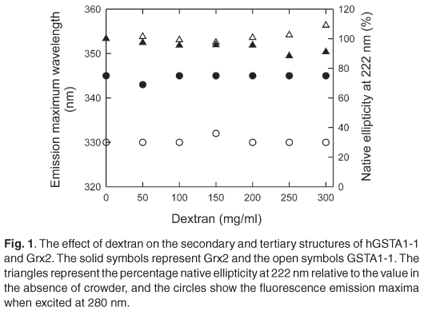
Urea denaturation experiments were performed using far-UV CD and tryptophan fluorescence to monitor structural changes that occur during isothermal unfolding, because both Grx2 and hGST A1-1 are denatured irreversibly by temperature. The unfolding of both proteins was performed at low protein concentrations (4 µM Grx2 and 0.5 µM hGST A1-1) in the absence and presence of 100 mg/ml dextran, as it is essential to use a system that displays a fully reversible unfolding equilibrium to study the effect of macromolecular crowding on the stability of proteins.2,13 Under these conditions, the recovery of the native state from urea-denatured protein was in excess of 80% with no observed aggregation. Higher concentrations of protein and/or dextran resulted in aggregation and significant losses in recoveries. Other studies have also shown that crowding often reduces refolding yields by causing aggregation during refolding.31-33
In the absence of crowder, urea-induced global unfolding of monomeric Grx2 and dimeric hGST A1-1 is two-state: N ↔ U for Grx2 (Gildenhuys, Wallace and Dirr, unpublished work) and N2* ↔ 2U for hGST A1-1,22 where N and U are the native and unfolded states, respectively, and N2* is hGST A1-1 with unfolded helix 9. This helix does not contribute towards the global stability of the protein.23 The equilibrium two-state unfolding mechanisms of these homologous proteins were preserved in the presence of 100 mg/ml dextran, as indicated by the monophasic and overlapping unfolding transitions obtained from CD and fluorescence data (Fig. 2). The coincident spectroscopic data demonstrate the simultaneous and cooperative loss of secondary and tertiary structures with increasing urea concentration. Furthermore, the transitions obtained from unfolding (forward) and refolding (reverse) experiments coincided, indicating the absence of hysteresis (data not shown). The continuous lines in Fig. 2 represent the best fit of the data to the two-state models for Grx2 and for hGST A1-1. The thermodynamic parameters of unfolding at 0 and 100 mg/ml dextran, as obtained from the fits, are reported in Table 1.
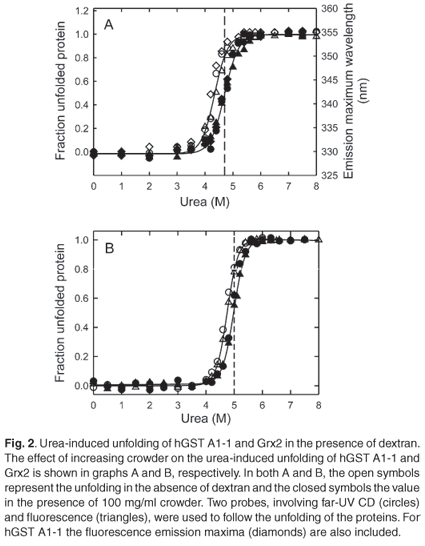

Dextran, at 100 mg/ml, shifts the unfolding transitions of Grx2 and hGST A1-1 to higher urea concentrations, as predicted by crowding theory.2 The Cm of both proteins (i.e. the urea concentration at the transition midpoint) is increased by about 0.3 M. The similar m-values (i.e. the dependence of ΔGN-D on urea or the unfolding cooperativity parameter) for the absence and presence of dextran (Table 1) is indicative of negligible specific intermolecular interactions between protein and crowder. At concentrations of urea where either protein is 80% unfolded in the absence of dextran (5 M urea for Grx2 and 4.7 M urea for hGST A1-1), the fraction of unfolded protein is reduced to 50% for both Grx2 and hGST A1-1 in the presence of 100 mg/ml dextran (Fig. 2). Compaction of the unfolded states of both proteins is indicated by an increase in alpha-helical content (i.e. increased negative ellipticity at 222 nm) and in the decreased solvent exposure of the tryptophan residues (i.e. blue shift in emission wavelength maximum). Given that the native states appear to be essentially unaffected by crowding, therefore, the dextran-induced formation of compact states of urea-denatured Grx2 and hGST A1-1 can be ascribed to steric excluded volume effects, which induce an entropic destabilization of expanded unfolded states.2,34 A similar effect has also been observed for the unfolded states of ribonuclease A14 and lysozyme.13
The difference between the values of ΔGN-D for the presence and absence of 100 mg/ml dextran indicate that the stability of Grx2 is increased by 1.1 kcal/mol, while that of hGST A1-1 is increased by 2.2 kcal/mol. Given the predicted linear dependence of the free energy of unfolding on the concentration of crowder,11-13 Grx2 and hGST A1-1 are stabilized by 0.011 and 0.022 kcal/mol, respectively, per g/l of dextran. The value for Grx2 is about 3-4 times larger than the corresponding values for other monomeric proteins11-13 but is similar to that for the molten globule state of cytochrome c.13 The ΔGN-D values 1.1 and 2.2 kcal/ mol correspond to a 7- and 44-fold reduction in the equilibrium-unfolding constant, KN-D, of Grx2 and hGST A1-1, respectively, in qualitative agreement with theoretical models.3 Unlike Grx2, the stability of hGST A1-1 is tightly coupled to the intrinsic stability of the individual subunits and the stabilizing interactions across the homodimer interface.35 The latter are proposed to contribute significantly towards stabilizing the tertiary structures of each subunit. Although crowding would influence both the folding and association of polypeptides, the contribution of the excluded volume effect to each process for hGST A1-1 is unclear. At this stage, theoretical models are not able to predict the extent to which oligomeric proteins will be stabilized by macromolecular crowding. According to the equivalent hard particle model for the excluded volume effect of dextran on the stability of monomeric proteins,13 the predicted dependence of ΔGN-D upon the concentration of crowder, mN-D, for a two-state unfolding process is:
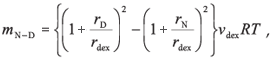
where rdex is the effective cylindrical radius of dextran [7 Å (ref. 36)]; νdex is the effective specific excluded volume of dextran [0.0008 l/g (ref. 37)]; rD and rN are the effective sphere radii of the unfolded and native states of a protein, respectively. The effective sphere radii of denatured (rD) and native (rN) Grx2 calculated from reff = (5/3)1/2Rg are 65 Å and 21 Å, respectively.3 The radii of gyration, Rg, were calculated according to Goldenberg,38 with Rg,D being the root-mean-square radius of gyration of the denatured state, taking long-range intramolecular steric interactions into account. The calculated mN-D value of 0.032 kcal/mol per g/l for Grx2 predicts that, at 100 mg/ml dextran, the protein should be stabilized by 3.2 kcal/mol. This, however, is about three times greater than the experimentally observed stabilizing effect. Realistic estimates of Rg,D are critical for predicting the effects of excluded volume on protein stability. Although we do not have experimental Rg,D data for Grx2, an excellent correlation between calculated and experimental values for several unfolded proteins has been observed.38 Furthermore, should the two cysteine residues in Grx2, Cys9 and Cys12, form a disulphide crosslink in the unfolded protein, in spite of the addition of DTT, the presence of the short loop in the unfolded polypeptide chain should not significantly influence its radius of gyration.38
There are currently limited experimental data to test the ability of theoretical models to predict the extent that excluded volume effects will stabilize globular proteins against unfolding by heat and denaturants. While some proteins are stabilized to an extent comparable to that predicted by theory,13,14 others are stabilized to a far lesser extent (this and other studies11,12). Much work is still required to develop and refine models for predicting reliably the effects of macromolecular crowding on protein stability, given the physicochemical complexity of this phenomenon. Further, it would be desirable to perform stability studies at concentrations of crowder that simulate crowding in cells but this may be hindered, as in this case, by impaired reversibility of unfolding and the formation of aggregates.
Effect of macromolecular crowding on GST activity
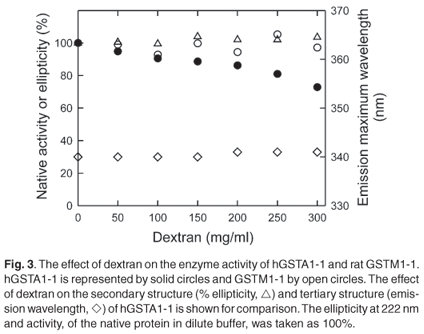
The polypeptide chain of each subunit of hGST A1-1 has an extended C-terminal region that forms an alpha helix (helix 9) over the active site.19,39 Because the dynamic behaviour of helix 9 plays an important role in the ligand binding and catalytic functions of the enzyme,39,40 enzyme activity has been used to probe perturbations in this region. Figure 3 shows that while the enzyme activity of hGST A1-1 decreases as the concentration of dextran increases, the activity of rGST M1-1 remains unaffected. The main difference between the two classes of GSTs is that M1-1 has a shorter C-terminal region and no helix 9. As dextran at 50-300 mg/ml does not impact on the structure and functionality of the M1-1 native state, macromolecular crowding appears not to affect the conformational dynamics of the enzyme, at least not of those regions involved in catalysis and product release which is rate-limiting for the substrate CDNB.41,42 The reduced activity of hGST A1-1 in the presence of dextran may, however, be due to a crowding-induced diminution in the conformational dynamics of helix 9. A less dynamic helix 9 has been shown to reduce enzyme activity with CDNB due to decreased substrate binding and product release, the latter being rate limiting.43 In addition, it has been reported that the activity of hGST A1-1 is reduced in cytosol, possibly via macromolecular crowding effects.44
Summary
In solution, proteins exist in equilibrium between their folded and unfolded states, the extent of which is determined by the stabilities of the individual states. Macromolecular crowding enhances the conformational stability of monomeric Grx2 and its homodimeric structural homologue hGST A1-1 by destabilizing their unfolded states, resulting in the equilibrium between native and unfolded states shifting towards the native state. Quantitatively, however, the extent of stabilization is less than that predicted by a theoretical hard particle model reported for the excluded volume effect on protein stability. Given the biological significance and wide-ranging effects of volume exclusion on protein stability and function, more experimental work is required to understand these effects better and for the development and refinement of predictive theoretical models.
This work was supported by the University of the Witwatersrand, the National Research Foundation (grant 205359), and the Research Chairs Initiative of the Department of Science and Technology and National Research Foundation (grant 64788). Any opinion, findings and conclusions or recommendations expressed in this material are exclusively those of the authors.
1. Ellis R.J. (2001). Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11, 114-119. [ Links ]
2. Minton A.P. (2000). Effect of a concentrated 'inert' macromolecular cosolute on the stability of a globular protein with respect to denaturation by heat and by chaotropes: a statistical-thermodynamic model. Biophys. J. 78, 101-109. [ Links ]
3. Minton A.P. (2005). Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: macromolecular crowding and protein stability revisited. Biophys. J. 88, 971-985. [ Links ]
4. Ghaemmaghami S. and Oas T.G. (2001). Quantitative protein stability measurement in vivo. Nature Struct. Biol. 8, 879-882. [ Links ]
5. Ignatova Z. and Gierasch L.M. (2004). Monitoring protein stability and aggregation in vivo by real-time fluorescent labeling. Proc. Natl Acad. Sci. USA 101, 523-528. [ Links ]
6. Martin J. and Hartl F.U. (1997). The effect of macromolecular crowding on chaperonin-mediated protein folding. Proc. Natl Acad. Sci. USA 94, 1107-1112. [ Links ]
7. Kinjo A.R. and Takada S. (2003). Competition between protein folding and aggregation with molecular chaperones in c rowded solutions: insight from mesoscopic simulations. Biophys. J. 85, 3521-3531. [ Links ]
8. Zhou H.X. (2004). Loops, linkages, rings, catenanes, cages, and crowders: entropy-based strategies for stabilizing proteins. Acc. Chem. Res. 37, 123-130. [ Links ]
9. Zhou H.X. (2004). Protein folding and binding in confined spaces and in crowded solutions. J. Mol. Recognit. 17, 368-375. [ Links ]
10. Cheung M.S., Klimov D. and Thirumalai D. (2005). Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc. Natl Acad. Sci. USA 102, 4753-4758. [ Links ]
11. Spencer D.S., Xu K., Logan T.M. and Zhou H.X. (2005). Effects of pH, salt, and macromolecular crowding on the stability of FK506-binding protein: an integrated experimental and theoretical study. J. Mol. Biol. 351, 219-232. [ Links ]
12. Qu Y. and Bolen D.W. (2002). Efficacy of macromolecular crowding in forcing proteins to fold. Biophys. Chem. 101-102, 155-165. [ Links ]
13. Sasahara K., McPhie P. and Minton A.P. (2003). Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol. 326, 1227-1237 [ Links ]
14. Tokuriki, N., Kinjo, M., Negi, S., Hoshino, M., Goto, Y., Urabe, I. and Yomo, T. (2004). Protein folding by the effects of macromolecular crowding. Protein Sci. 13, 125-133. [ Links ]
15. Bolis D., Politou A.S., Kelly G., Pastore A. and Temussi P.A. (2004). Protein stability in nanocages: a novel approach for influencing protein stability by molecular confinement. J. Mol. Biol. 336, 203-212. [ Links ]
16. Eggers D.K. and Valentine J.S. (2001). Molecular confinement influences protein structure and enhances thermal protein stability. Protein Sci. 10, 250- 261. [ Links ]
17. Campanini, B., Bologna, S., Cannone, F., Chirico, G., Mozzarelli, A., and Bettati, S. (2005). Unfolding of Green Fluorescent Protein mut2 in wet nanoporous silica gels. Protein Sci. 14, 1125-1133. [ Links ]
18. Myers J.K., Pace C.N. and Scholtz J.M. (1995). Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4, 2138-2148. [ Links ]
19. Sinning I., Kleywegt G.J., Cowan S.W., Reinemer P., Dirr H.W., Huber R., Gilliland G.L., Armstrong R.N., Ji X., Board P.G., Olin B., Mannervik B. and Jones T.A. (1993). Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J. Mol. Biol. 232, 192-212. [ Links ]
20. Xia B., Vlamis-Gardikas A., Holmgren A., Wright P.E. and Dyson H.J. (2001). Solution structure of Escherichia coli glutaredoxin-2 shows similarity to mammalian glutathione-S-transferases. J. Mol. Biol. 310, 907-918. [ Links ]
21. Vlamis-Gardikas A., Aslund F., Spyrou G., Bergman T. and Holmgren A. (1997). Cloning, overexpression, and characterization of glutaredoxin 2, an atypical glutaredoxin from Escherichia coli. J. Biol. Chem. 272, 11236-11243. [ Links ]
22. Wallace L.A., Sluis-Cremer N. and Dirr H.W. (1998). Equilibrium and kinetic unfolding properties of dimeric human glutathione transferase A1-1. Biochemistry 37, 5320-5328. [ Links ]
23. Dirr H.W. and Wallace L.A. (1999). Role of the C-terminal helix 9 in the stability and ligandin function of class alpha glutathione transferase A1-1. Biochemistry 38, 15631-15640. [ Links ]
24. Stenberg G., Bjornestedt R. and Mannervik B. (1992). Heterologous expression of recombinant human glutathione transferase A1-1 from a hepatoma cell line. Protein Expr. Purif. 3, 80-84. [ Links ]
25. Xia B., Chung J., Vlamis-Gardikas A., Holmgren A., Wright P.E. and Dyson H.J. (1999). Assignment of 1H, 13C, and 15N resonances of reduced Escherichia coli glutaredoxin 2. J. Biomol. NMR 14, 197-198. [ Links ]
26. Sayed Y., Wallace L.A. and Dirr H.W. (2000). The hydrophobic lock-and-key intersubunit motif of glutathione transferase A1-1: implications for catalysis, ligandin function and stability. FEBS Lett. 465, 169-172. [ Links ]
27. Hornby J.A.T., Luo J-K., Stevens J.M., Wallace L.A. Kaplan W. Armstrong R.N. and Dirr H.W. (2000). Equilibrium folding of dimeric class µ glutathione transferases involves a stable monomeric intermediate. Biochemistry 39, 12336-12344. [ Links ]
28. Habig W.H. and Jakoby W.B. (1981). Assays for differentiation of glutathione S-transferases. Methods Enzymol. 77, 398-405. [ Links ]
29. Laurent T.C. (1963). The interaction between polysaccharides and other macromolecules. Biochem. J. 89, 253-257. [ Links ]
30. Laurent T.C. and Ogston A.G. (1963). The interaction between polysaccharides and other macromolecules. 4. The osmotic pressure of mixtures of serum albumin and hyaluronic acid. Biochem. J. 89, 249-253. [ Links ]
31. van den Berg B., Ellis R.J. and Dobson C.M. (1999). Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 18, 6927-6933. [ Links ]
32. Li J., Zhang S. and Wang C. (2001). Effects of macromolecular crowding on the refolding of glucose-6-phosphate dehydrogenase and protein disulfide isomerase. J. Biol. Chem. 276, 34396-34401. [ Links ]
33. Fernandez A. and las Mercedes B.M. (2002). Solvent environment conducive to protein aggregation. FEBS Lett. 529, 298-302. [ Links ]
34. Minton A.P. (2001). The influence of macromolecular crowding and macromolecular confinement on biochemical reacions in physiological media. J. Biol. Chem. 276, 10577-10580. [ Links ]
35. Wallace L.A., Burke J. and Dirr H.W. (2000). Domain-domain interface packing at conserved Trp-20 in class alpha glutathione transferase impacts on protein stability. Biochim. Biophys. Acta 1478, 325-332. [ Links ]
36. Laurent T.C. and Killander J. (1964). A theory of gel filtration and its experimental verification. J. Chromatography 14, 317-330. [ Links ]
37. Rivas G., Fernandez J.A. and Minton A.P. (1999). Direct observation of the self-association of dilute proteins in the presence of inert macromolecules at high concentration via tracer sedimentation equilibrium: theory, experiment, and biological significance. Biochemistry 38, 9379-9388. [ Links ]
38. Goldenberg D.P. (2003). Computational simulation of the statistical properties of unfolded proteins. J. Mol. Biol. 326, 1615-1633. [ Links ]
39. Cameron A.D., Sinning I., L'Hermite G., Olin B. Board P.G., Mannervik B. and Jones T.A. (1995). Structural analysis of human alpha-class glutathione transferase A1-1 in the apo-form and in complexes with ethacrynic acid and its glutathione conjugate. Structure 3, 717-727. [ Links ]
40. Allardyce C.S., McDonagh P.D., Lian L.Y., Wolf C.R. and Roberts G.C. (1999). The role of tyrosine-9 and the C-terminal helix in the catalytic mechanism of Alpha-class glutathione S-transferases. Biochem. J. 343, 525-531. [ Links ]
41. Codreanu S.G., Ladner J.E., Xiao G., Stourman N.V., Hachey D.L., Gilliland G.L. and Armstrong R.N. (2002). Local protein dynamics and catalysis: detection of segmental motion associated with rate-limiting product release by a glutathione transferase. Biochemistry 41, 15161-15172. [ Links ]
42. Codreanu S.G., Thompson L.C., Hachey D.L., Dirr H.W. and Armstrong R.N. (2005). Influence of the dimer interface on glutathione transferase structure and dynamics revealed by amide H/D exchange mass spectrometry. Biochemistry 44, 10605-10612. [ Links ]
43. Nilsson L.O., Edalat M., Pettersson P.L. and Mannervik B. (2002). Aromatic residues in the C-terminal region of glutathione transferase A1-1 influence rate-determining steps in the catalytic mechanism. Biochim. Biophys. Acta 1597, 157-163. [ Links ]
44. Sundberg K., Dreij K., Seidel A. and Jernstrom B. (2002). Glutathione conjugation and DNA adduct formation of dibenzo[a,l]pyrene and benzo[a]pyrene diol epoxides in V79 cells stably expressing different human glutathione transferases. Chem. Res. Toxicol. 15, 170-179. [ Links ]
45. Pace C.N. (1986). Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131, 266-280. [ Links ]
Received 11 July. Accepted 26 October 2007.
Abbreviations used: CDNB, 1-chloro-2,4-dinitrobenzene; Grx2, glutaredoxin2; GST, glutathione transferase; hGST A1-1, homodimeric human class Alpha GST with two type 1 subunits; rGST M1-1, homodimeric rat class Mu GST with two type 1 subunits.
Author for correspondence. E-mail: heinid@gecko.biol.wits.ac.za
Materials
Dextran 70 (clinical grade) was from Sigma (St Louis, Missouri). Ultrapure urea was from Merck (Darmstadt, Germany). DTT was obtained from Whitehead Scientific (Cape Town) and GSH was from ICN Biomedicals (Aurora, Ohio). All other reagents were of analytical grade. The pKHA1 plasmid that encodes hGST A1-1 and the pET24a plasmid that encodes Grx2 were gifts from B. Mannervik (Department of Biochemistry, University of Uppsala, Sweden)24 and J. Dyson (The Scripps Research Institute, California),25 respectively.
Protein expression and purification
Human GST A1-1 was overexpressed in BL21 Escherichia coli cells containing the pKHA1 plasmid, purified by CM-Sephadex chromatography,26 stored in 20 mM sodium phosphate, 1 mM EDTA, 0.02% sodium azide, pH 6.5. Grx2 was overexpressed in E. coli BL21(DE3)pLys S cells containing the pET24a plasmid vector, purified by DEAE-Sepharose anion-exchange chromatography,21,25 and stored in the same buffer as for hGST A1-1 but with 1 mM DTT. Rat GST M1-1 was overexpressed in E. coli M5219 transformed with pGT33MX and purified by CM-Sephadex chromatography, and stored in 20 mM sodium phosphate buffer, pH 6.5, with 0.1 M NaCl and 0.02% sodium azide.27 The purity of the proteins was assessed by SDS-PAGE and SEC-HPLC and the concentrations of hGST A1-1, Grx2 and rGST M1-1 were determined spectrophotometrically at 280 nm using extinction coefficients of 38 200 M-1cm-1, 21 860 M-1cm-1 and 81 480 M-1cm-1, respectively.
Unfolding studies
All of the unfolding experiments were performed at 20°C in 20 mM sodium phosphate, 1 mM EDTA, 0.02% sodium azide, pH 6.5 for hGST A1-1 and at pH 7 in the presence of 1 mM DTT for Grx2. Urea-induced unfolding was performed by incubating native protein with increasing concentrations of urea (0-8 M), in the absence or presence of dextran. The final dimeric hGST A1-1 concentration was 0.5 µM and for Grx2 a final monomeric concentration of 4 µM was used. Structural changes were monitored by far-UV CD at 222 nm and tryptophan fluorescence. Far-UV CD measurements were made in a Jasco model J-810 CD spectropolarimeter at 20°C using a 1-mm pathlength cuvette. Spectra were an average of 15 scans. The intrinsic tryptophan fluorescence of the proteins, excited at 280 nm, was measured with a Perkin Elmer luminescence spectrometer model LS 50B. Excitation at 280 nm enhances the signal of tryptophan fluorescence due to the transfer of excitation energy from tyrosine residues to tryptophan residues. The change in fluorescence of Grx2 was monitored at a single wavelength of 345 nm, the peak emission wavelength of the folded protein. For hGST A1-1, the extent of unfolding was determined by the ratio of the fluorescence intensity at 355 nm (unfolded protein) to the intensity at 330 nm (folded protein). The unfolding data were analysed by non-linear regression using two-state models for both proteins.
GST activity assays
The enzyme activity was measured spectroscopically at 340 nm by monitoring the formation of S-2,4-dinitrophenyl glutathione in 0.1 M sodium phosphate, 1 mM EDTA, pH 6.5, containing 1 mM glutathione and 1 mM CDNB28 in the absence or presence of dextran 70 (50-300 mg/ml).

