










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.104 n.1-2 Pretoria Jan./Feb. 2008
RESEARCH IN ACTION
Protein homology modelling and its use in South Africa
A. Ozlem Tastan Bishop; Tjaart A. P. de Beer; Fourie Joubert
Bioinformatics and Computational Biology Unit, Department of Biochemistry, University of Pretoria, Pretoria 0002, South Africa
ABSTRACT
Homology modelling is an important computational technique, within structural biology, to determine the 3D structure of proteins. It uses available high-resolution protein structures to produce a model of a protein of similar, but unknown, structure. We describe the essential steps in the process, and discuss the circumstances in which homology modelling is likely to lead to a useful result. Homology modelling plays a valuable role in drug design, and we illustrate this by one example, anti-SARS inhibitors. In South Africa, homology modelling has been applied to proteins that may be relevant for drug design in connection with diseases as well as in other potential industrial applications. The use to date has been limited, however, so this article aims to introduce this useful and cost-effective technique to a wider community.
Introduction
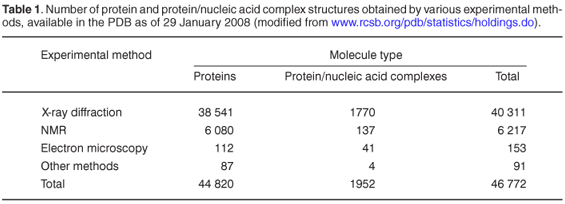
It is almost 50 years since the first proteinrystal structure, of myglobin, was solved.1,2 With advances in experimental structural biology techniques as well as in whole-genome sequencing in the late 1990s, the excitement of solving a single-crystal structure has been replaced by determining protein structures on a large scale. This was the beginning of a new field, structural genomics, with active centres around the world.3-6 The contribution of structural genomics to the Protein Data Bank (PDB, an electronic repository of 3D structures of proteins and nucleic acids) is considerable, and nowadays these initiatives contribute approximately half of the new structurally characterized families of proteins.7
There are currently (January 2008) 44 820 protein structures in the PDB (Table 1). Although this number is increasing rapidly, there remains a huge gap between the number of available gene sequences and experimentally solved protein structures. X-ray crystallography, NMR and electron microscopy proceed much more slowly than genome sequencing; and since many more genome sequences are on the way, this gap must surely grow. So, what could be the solution? Structural genomics projects aim to determine experimentally at least one representative 3D structure for every protein family, and then to exploitthe fact that proteins from the same familyare evolutionarily related and share similar sequences and structures.8 This means that instead of trying to characterize the structure of every protein experimentally, we may start from known representative structures and use computational methods to predict the structure of related proteins. This method has become known as homology modelling. According to the New York Structural Genomics Research Consortium, for each new structure, on average about 100 protein sequences without any prior structural characterization could be modelled at least at the fold level.9
Why is there this great effort to solve protein structures? Because they carry large amounts of information, and influence drug discovery, as one example of an application, at every stage in the design process.10,11 HIV/AIDS drugs such as Agenerase and Viracept were developed using the crystal structure of HIV protease.12,13 Thus, no matter how obtained (experimentally, computationally or using both approaches), 3D protein structure is of undeniable importance.
Structural biology in South Africa is a relatively new field that cannot yet be regarded as fully established. In this review, we discuss one of the techniques, homology modelling, and its applications. Our aim is to familiarize more research groups in biology with homology modelling, as it has great potential and can be learnt relatively easily and quickly.
Steps in homology modelling
Homology modelling seeks to predict the 3D structure of a protein based on its sequence similarity to one or more proteins of known structure. The method relies on the observation that the structural conformation of a protein is more highly conserved than its amino acid sequence. Homology modelling can be divided into four steps: template identification, alignment, model building and refinement, and validation (Fig. 1), with various computational tools (Table 2) available for each step. More detailed information is available in recently published reviews.10-16
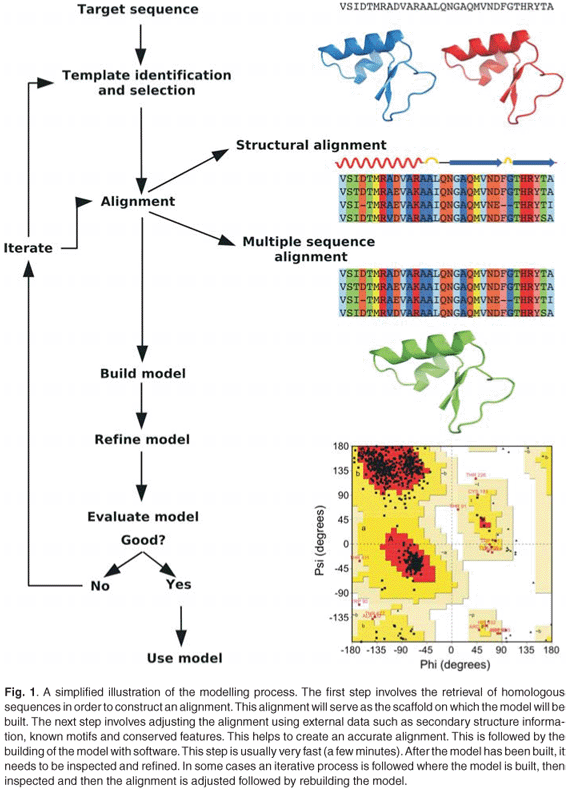
Template identification
Template identification is the critical first step. It lays the foundation by identifying appropriate homologue(s) of known protein structure, called template(s), which are sufficiently similar to the target sequence to be modelled. A simple search submits the target sequence to programs such as BLAST17 or FASTA.18 However, these programs work well only for alignment of sequences with high similarities. Methods such as PSI-BLAST19 and ScanPS20 have recently increased the possibility of detecting distant homologues.
These methods often suggest several candidate templates. The ideal is to identify the template(s) which has the highest percentage identity to the target, has the highest resolution, and has structures with (or without) appropriate ligands and/or cofactors. It may be that there is no candidate template that is best according to all criteria, in which case the choice is a matter of judgment and perhaps of trying different templates.
Alignment
The next step involves creating an alignment of the target sequence with the template structure(s). This is a vital step and there are various ways to ensure high accuracy. The target and template sequence can be aligned with a protein domain family alignment retrieved from Pfam,21 or a custom alignment can be generated from all relevant sequences retrieved via BLAST. Programs such as Clustal,22 Muscle,23 and TCoffee24 can be used to construct the alignment. Sometimes structural alignments are preferred, especially for distantly related sequences, because structure is more conserved than sequence.3DCoffee,25 FUGUE26 and mGenThreader27 are well-known structural alignment programs. MEME28 provides information about conserved motifs found in aligned sequences, and can be used to guide the alignment.
The alignment can and should be optimized manually. By including biological information such as the solvation environment of an amino acid, better-informed changes to the alignment can be made by the user. This type of information is not often available to the alignment program.
Model building and refinement
Although the theory behind building a protein homology model is complicated, using available programs is relatively easy. Several modelling programs are available, using different methods to construct the 3D structures. In segment matching methods, the target is divided into short segments, and alignment is done over segments rather than over the entire protein.29 Satisfying spatial restraintsis the most common method. It uses either distances or optimization techniques to satisfy the spatial restraints. The method is implemented using the popular program, Modeller,30 which includes the CHARMM31 energy terms that ensure valid stereochemistry is combined with spatial restraints. There are several stand-alone modelling programs available such as WHAT IF.32 Web servers such as SwissModel and the Rosetta server make it even easier to generate a model.
A problem regularly encountered in homology modelling is loops. A general guideline is that any insertion/loop longer than about five residues should be omitted. There are programs which try to model loops such as Modeller and its more specialized loop modelling server, Modloop.33
The initial model may have suboptimal bond angles and lengths. Such deficiencies can be adjusted by an energy minimization procedure, but a difficulty is that it moves atoms towards a local minimum, which may not be a global minimum over all possible conformations. Alternatively, molecular dynamics can be used, in which the motion of the whole protein is modelled.34,35
Validation
After being built, the model needs to be validated. One of the most thorough structure checking programs is Whatcheck.36 Other programs such as Procheck,37 and ANOLEA38 at SwissModel evaluate fewer parameters. The best validation combines common sense, biological knowledge and results from analytical tools. Some models will need further refinement. There is a cycle between building-validating-refining. Most refinement involves adjusting the alignment.
Advantages and limitations of homology modelling
Homology modelling is a relatively easy technique. It takes much less time to learn, to do the calculations and obtain a result, than an experiment. Nor does it require expensive experimental facilities, just a standard desktop computer. In the absence of high-resolution experimental structures, therefore, homology modelling can be of much value.
However, the quality and accuracy of the homology model depend on several factors. The technique requires a high-resolution experimental protein structure as a template, the accuracy of which directly affects the quality of the model. Even more importantly, the quality of the model depends on the degree of sequence identity between the template and protein to be modelled.9,10,39-41 Alignment errors increase rapidly when the sequence identity is less than 30%. Mediumaccuracy homology models have between about 30% and 50% sequence identity to the template. They can facilitate structure-based prediction of target for 'drugability', the design of mutagenesis experiments and the construction of in vitro test assays. Higher accuracy models are typically obtained when there is more than 50% sequence identity. They can be used in the estimation of protein-ligand interactions, such as the prediction of the preferred sites of metabolism of small molecules, as well as structure-based drug design.
Homology modelling of membrane proteins requires particular care. The available crystal structures are limited, and modelling methods are mainly designed for water-soluble proteins. Comparing results from different methods is one approach.42 Another limitation of homology modelling is the presence of loops and inserts, as they cannot be modelled without template data; however, one can still estimate length, location, and distance from the active site if the protein is an enzyme.
Applications of homology modelling
There is much information concerning biological function that can be derived from a 3D protein structure.43 The residues that are buried in the core of the molecule or exposed to solvent on its surface can be identified. Protein-ligand complexes carry functional information such as where the ligand is bound, and, if the protein is an enzyme, which residues in the active site interact with the ligand. Protein structures can also be used to explain the effects of mutations in drug resistance and in genetic diseases.44,45 Analysis of a protein structure and function generally has many applications, from basic mutagenesis experiments to various stages of the drug discovery process.
Here we give just one example of a breakthrough in drug design that used homology modelling. Severe acute respiratory syndrome (SARS) was identified in China in 2002 and quickly spread to other countries. The cause was a new coronavirus (CoV). Soon afterwards, whole genomes of different SARS-CoV strains were solved. Main protease (Mpro), which has an important role in virus replication, became an immediate drug target. CoV-Mpro has 40% and 46% sequence identity to transmissible gastroenteritis coronavirus (TGEV) Mpro, and human coronavirus 229E, respectively, and X-ray structures were already available. Several groups released the homology model of the protease in May 2003.46-48 A comparison of the inhibitor complexed with TGEV-Mpro with available inhibitor complexes in PDB gave a similar inhibitor- binding mode in the complex of human rhino-virus type 2 (HRV2) 3C proteinase with AG7088. At the time, AG7088 was in clinical trials for the treatment of the human rhino-virus that causes the common cold. AG7088 was docked into the substrate-binding site of the SARS-CoV- Mpro model, indicating that it would be a good starting point for the design of anti-SARS drugs.47 Shortly thereafter, it was shown that AG7088 does indeed have anti-SARS activity in vitro.
Protein homology modelling in South Africa
Homology modelling in South Africa has involved projects on disease-related proteins as well as on proteins with industrial applications. We highlight some of these below.
Sleeping sickness is a severe disease in Africa, caused by trypanosomas. There are problems with current drugs such as their high toxicity and ineffectiveness due to drug resistance. The enzyme, glycerol-3-phosphate dehydrogenase (GPDH), from the glycolytic metabolic pathway of Leishmania mexicana, has been elucidated as a possible new drug target.49 At Rhodes University, this information, together with the availability of a homologue crystal structure of Leishmania mexicana, led to the construction of a homology model of NAD-dependent glycerol-3-phosphate dehydrogenase of Trypasonoma brucei rhodesiense.50 The model was further used in molecular dynamic simulations to show that the protein can be used for drug design.
Ticks transmit pathogens and toxic compounds, severely affecting the health of the host, even leading to death. The development of tick control methods requires an understanding of tick-host interactions. A group at the University of Pretoria used homology modelling to study the structure of a thrombin inhibitor, savignin, from the tick, Ornithodoros savignyi.51 The aim was to understand the anti-clotting function of savignin, with the long-term goal of the design of anti-haemostatic pharmaceuticals.
Malaria affects 300-500 million people annually and kills approximately 2 million.52 Plasmodium falciparum is the most lethal malaria parasite infecting humans, and the emergence of drug-resistant strains demands a search for new targets. This depends on structural information. However, there are difficulties with malaria proteins. Crystallization trials are limited due to the difficulty of expressing malaria proteins in high yield and pure state, as well as by the presence of long and disordered inserts. These Plasmodium-specific inserts also make it difficult to construct homology models. A group at the University of Pretoria has computed homology models of malaria proteins. In the polyamine pathway, models of ornithine decarboxylase (ODC)53 and S-adenosylmethionine decarboxylase54 were prepared. The comparison of these models with the human enzyme showed differences in the active site, which might allow identification of parasite-specific inhibitors. P. falciparum spermidine synthase (PfSpdSyn) is another enzyme in this pathway. Molecular dynamics of the homology model demonstrated the mechanism of the aminopropyltransferase action of PfSpdSyn.55
The folate biosynthetic pathway is also important for drug targeting. Homology models of the bifunctional enzymes hydroxymethylpterin pyrophosphokinase-dihydropteroate synthase (PPPK- DHPS) were constructed. The aim was to investigate possible causes of the development of resistance in mutations.56 In the vitamin B6 system, an homology model of pyridoxal kinase (PdxK) showed the presence of a parasite-specific insert (roughly 200 residues) and a number of differences between the eukaryotic and malarial enzyme,57 which might lead to the identification of parasite-specific inhibitors. Thus, several approaches and proteins are being investigated with a view to designing new drugs against malaria.
Researchers at the University of Cape Town are working on nitrilase, an industrial enzyme. Nitrilases convert nitriles to the corresponding acids and ammonia. They are used to manufacture the biologically active enantiomers such as (R)-mandelic acid, (S)-phenyl-lactic acid, and (R)-3-hydroxy-4-cyano-butyric acid, which are key intermediates in the synthesis of the anti-cholesterol drug Lipitor®.58 The group determined the low- resolution structures of two cyanide-degrading members of this family from Pseudomonas stutzeri (CynDstu) and Bacillus pumilus (CynDpum) by electron microscopy.59,60 They found that they have spiral structures with 14 and 18 subunits, respectively, unlike distant homologues that have dimeric structures. By resolving the structures of these distant homologues at an atomic level, the group was able to model the structures and interpret their low-resolution data. The combination of homology modelling and electron microscopy results enabled them to identify the interfaces that lead to spiral oligomer formation and to postulate which residues are involved in interactions across the interface. Later, the UCT group applied the same idea to another nitrilase, from Rhodococcus rhodochrous J1, to study interaction surfaces.61 Overall, they showed the relationship between the formation of oligomers and the activity of these enzymes, and in the long term the work is expected to have direct relevance for various biotechnological application of nitrilase enzymes.
Researchers at the University of the Western Cape are interested in another industrial enzyme, nitrile hydratase (NHase), which catalyses the conversion of nitriles to their corresponding amides. This enzyme has been the subject of interest to both academics and industry for over two decades, mostly due to its biocatalytic activity (in acrylamide and nicotinamide production). Its importance extends also to its use for environmental remediation by removing nitriles from waste streams. The group at UWC generated the homology model of the enzyme from thermophile Bacillus pallidus RAPc8 as well as a 122-amino-acid accessory protein involved in thermostable NHase expression (P14K), a homologue of the 2Fe-2S class of ferredoxins.62 Modelling of the P14K protein structure suggested that the protein functions as a subunit-specific chaperone, and helps with the folding of the NHase and the formation of NHase heterotetramer.
Lipases are a widely used group of biocatalysts,63 and can be produced in large quantities from fungi and bacteria. They do not require cofactors nor do they catalyse side reactions, and are thus very attractive for industrial applications. They hydrolyze triacylglycerols to fatty acids and acylglycerols. Lipases are used for the synthesis of biopolymers, biodiesel, and the production of enantiopure pharmaceuticals, agrochemicals, cosmetics and flavour compounds. A group from the University of the Free State investigated an extracellular lipase from Pseudomonas luteola.64 Homology modelling of this lipase enabled them to compare it with the other available lipase structures, concluding that this enzyme could also have biocatalytic applications.
Conclusion
We have given an overview of what homology modelling is all about: procedure, applications, advantages and limitations, as well as its current use in South Africa. Homology modelling is entirely a computational process and much easier to implement than the experimental path to structural information about a protein, although it relies on suitable experimental structures being already known. Applications of homology modelling can range from design of the next experiment in an ongoing biochemical investigation, to the discovery of drugs with important disease control properties. On the other hand, in circumstances where the homology model may be of only limited accuracy, the results may require experimental verification.
Because homology modelling has been used in South Africa in only a limited way, we hope that this article will introduce a broader community to this relatively simple and cost-effective technique, and so lead to practical applications, especially in tackling our own problems—of HIV/ AIDS, tuberculosis and malaria, for example—rather than waiting for solutions from elsewhere.
A.Ö.T.B. thanks the Claude Leon Foundation and University of Pretoria for financial support. T.D.B. thanks the National Bioinformatics Network for a Ph.D. bursary.
1. Kendrew J.C., Bodo G., Dinitzis H.M., Parrish R.G., Wyckoff H. and Phillips D.C. (1958). A three-dimensional model of the myglobin molecule obtained by X-ray analysis. Nature 181, 662- 666. [ Links ]
2. Kendrew J.C. (1961). The three-dimensional structure of a protein molecule. Sci. Am. 205(6), 96-110. [ Links ]
3. Cassman M. and Norvell J.C. (1999). Support for structural genomics and synchrotrons. Science 286, 239-240. [ Links ]
4. Yokoyama S., Hirota H., Kigawa T., Yabuki T., Shirouzu M., Terada T., Ito Y., Matsuo Y., Kuroda Y., Nishimura Y., Kyogoku Y., Miki K., Masui R. and Kuramitsu S. (2000). Structural genomics projects in Japan. Nature Struct. Biol. Suppl. 943-945. [ Links ]
5. Bahar M., Ballard C., Coen S.X., Cowtan K.D., Dodson E.J., Emsley P., Esnouf R.M., Keegan R. et al. (2006). SPINE workshop on automated X-ray analysis: a progress report. Acta Cryst. D Biol. Crystallogr. 62, 1170-1183. [ Links ]
6. Williamson A.R. (2000). Creating a structural genomics consortium. Nature Struct. Biol. Suppl. 953. [ Links ]
7. Marsden R.L., Lewis T.A. and Orengo C.A. (2007). Towards a comprehensive structural coverage of completed genomes: a structural genomics viewpoint. BMC Bioinformatics 8, 86. [ Links ]
8. Sander C. and Schneider R. (1991). Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 9, 56-68. [ Links ]
9. Baker D. and Sali A. (2001). Protein structure prediction and structural genomics. Science 294, 93-96. [ Links ]
10. Hillisch A., Pineda L.F. and Hilgenfeld R. (2004). Utility of homology models in the drug discovery process. Drug Discov. Today 9, 659-669. [ Links ]
11. Blundell T.L., Sibanda B.L., Montalvao R.W., Brewerton S., Chelliah V., Worth C.L., Harmer N.J., Davies O. and Burke O. (2006). Structural biology and bioinformatics in drug design: opportunities and challenges for target identification and lead discovery. Phil. Trans. R. Soc. B 361, 413-423. [ Links ]
12. Lapatto R., Blundell T., Hemmings A., Overington J., Wilderspin A., Wood S., Merson J.R., Whittle P.J., Danley D.E., Geoghegan K.F. et al. (1989). X-ray analysis of HIV-1 proteinase at 2.7A resolution confirms structural homology among retroviral enzymes. Nature 342, 299-302. [ Links ]
13. Miller M., Schneider J., Sathyanarayana B.K., Toth M.V., Marshall G.R., Clawson L., Selk L., Kent S.B. and Wlodawer A. (1989). Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 Å resolution. Science 246, 1149- 1152. [ Links ]
14. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V.A., Pieper U., Stuart A.C., Marti-Renom M.A., Madhusudhan M.S., Yerkovich B. and Sali A. (2003). Nucl. Acids Res. 31, 3375-3380. [ Links ]
15. Dunbrack R.L. Jr (2006). Sequence comparison and protein structure prediction. Curr. Opin. Struct. Biol. 16, 374-384. [ Links ]
16. Xiang Z. (2006). Advances in homology protein structure modeling. Curr. Protein Pept. Sci. 7, 217-227. [ Links ]
17. Altschul S.F., Gish W., Miller W., Myers E.W. and Lipman D.J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403-410. [ Links ]
18. Pearson W.R. (1990). Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 183, 63-98. [ Links ]
19. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W. and Lipman D.J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 25, 3389-3402. [ Links ]
20. Barton G.J. (1992). Computer speed and sequence comparison. Science 257, 1609-1610. [ Links ]
21. Sonhammer E.L., Eddy S.R. and Durbin R. (1997). Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins 28, 405-420. [ Links ]
22. Higgins D.G. and Sharp P.M. (1988). CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene 73, 237-244. [ Links ]
23. Edgar R.C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. [ Links ]
24. Notredame C., Higgins D. and Heringa J. (2000). T-Coffee: a novel method for multiple sequence alignments. J. Mol. Biol. 302, 205-217. [ Links ]
25. O'Sullivan O., Suhre K., Abergel C., Higgins D.G. and Notredame C. (2004). 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J. Mol. Biol. 340, 385-395 [ Links ]
26. Shi J., Blundell T.L. and Mizuguchi K. (2001). FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310, 243-257. [ Links ]
27. McGuffin L.J. and Jones D.T. (2003). Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics 19, 874-881. [ Links ]
28. Bailey T.L. and Elkan C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Second International Conference on Intelligent Systems for Molecular Biology, pp. 28-36. AAAI Press, Menlo Park, CA. [ Links ]
29. Levitt M. (1992). Accurate modelling of protein conformation by automatic segment matching. J. Mol. Biol. 226, 507-533. [ Links ]
30. Sali A. and Blundell T.L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779-815. [ Links ]
31. Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. and Karplus M. (1983). CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4, 187-217. [ Links ]
32. Vriend G. (1990). WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8, 52-56. [ Links ]
33. Fiser A., Do R.K. and Sali A. (2000). Modeling of loops in protein structures. Protein Science 9, 1753-73. [ Links ]
34. Krieger E., Nabuurs S.B. and Vriend G. (2003). Homology modeling. In Structural Bioinformatics, pp. 507-521. Wiley-Liss, New York. [ Links ]
35. Xiong J. (2006). Protein tertiary structure prediction. In Essential Bioinformatics, pp. 214-230. Cambridge University Press, Cambridge. [ Links ]
36. Hooft R.W.W., Vriend G., Sander C. and Abola E.E. (1996). Errors in protein structures. Nature 381, 272-272. [ Links ]
37. Laskowski R.A., MacArthur M.W., Moss D.S. and Thornton J.M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283-291. [ Links ]
38. Melo F., Devos D., Depieruex E. and Feytmans E. (1997). ANOLEA: a www server to assess protein structures. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5, 187-190. [ Links ]
39. Marti-Renom M.A., Stuart A.C., Fiser A., Sanchez R., Melo F. and Sali A. (2000). Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291-325. [ Links ]
40. Sanchez R. and Sali A. (1998). Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc. Natl Acad. Sci. USA 95, 13597- 13602. [ Links ]
41. Koehl P. and Levitt M. (1999). A brighter future for protein structure prediction. Nature Struct. Biol. 6, 108-111. [ Links ]
42. Reddy Ch.S., Vijayasarathy K., Srivinas E., Sastry G.M. and Sastry G.N. (2006). Homology modeling of membrane proteins: a critical assessment. Comput. Biol. Chem. 30, 120-126. [ Links ]
43. Thornton J.M., Todd A.E., Milburn D., Borkakoti N. and Orengo C.A. (2000). From structure to function: approaches and limitations. Nature Struct. Biol. Suppl. 991-994. [ Links ]
44. Marbotti A. and Facchiano A.M. (2005). Homology modeling studies on human galactose-1-phosphate uridylytransferase and on its galactosemia-related mutant Q188R provide an explanation of molecular effects of the mutation on homo- and heterodimers. J. Med. Chem. 48, 773-779. [ Links ]
45. Rempel B.P., Clarke L.A. and Withers S.G. (2005). A homology model for human α-L-iduronidase: insights into human disease. Mol. Genet. Metab. 85, 28-37. [ Links ]
46. Takeda-Shitaka M., Takaya D., Chiba C., Tanaka H. and Umeyama H. (2004). Protein structure prediction in structure based drug design. Curr. Med. Chem. 11, 551-558. [ Links ]
47. Anand K., Ziebuhr J., Wadhwani P., Mesters J.R. and Hilgenfeld R. (2003). Coronavirus main proteinase (3CLpr) structure: basis for design of anti-SARS drugs. Science 300, 1763-1767. [ Links ]
48. Xiong B., Gui C.S., Xu X.Y., Luo C., Chen J., Luo H.B., Chen L.L., Li G.W., Sun T., Yu C.Y, Yue L.D. et al. (2003). A 3D model of SARS-CoV3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sinica 24, 497-504. [ Links ]
49. Suresh S., Turley S., Opperdoes F.R., Michels P.A. and Hol W.G. (2000). A potential target enzyme for trypanocidal drugs revealed by the crystal structure of NAD-dependent glycerol-3-phosphate dehydrogenase from Leishmania mexicana. Structure 8, 541-552. [ Links ]
50. Zubrzycki I.Z. (2002). Homology modeling and molecular dynamics study of NAD-dependent glycerol-3-phosphate dehydrogenase from Trypanosoma brucei rhodesiense, a potential target enzyme for anti-sleeping sickness drug development. Biophys. J. 82, 2906-2915. [ Links ]
51. Mans B.J., Louw A.I and Neitz A.W.H. (2002). Amino acid sequence and structure modeling of savinin, a thrombin inhibitor from the tick, Ornithodoros savignyi. Insect Biochem. Mol. Biol. 32, 821-828. [ Links ]
52. Breman J. (2001). The ears of the hippopotamus: manifestations, determinants, and estimates of the malaria burden. Am. J. Trop. Med. Hyg. 64, 1-11. [ Links ]
53. Birkholtz L., Joubert F., Neitz A.W.H. and Louw A.I. (2003). Comparative properties of a three-dimensional model of Plasmodium falciparum ornithine decarboxylase. Proteins 50, 464-473. [ Links ]
54. Wells G.A., Birkholtz L.M., Joubert F., Walter R.D. and Louw A.I. (2006). Novel properties of malarial S-adenosylmethionine decarboxylase as revealed by structural modeling. J. Mol. Graph. Model. 24, 307-318. [ Links ]
55. Burger P.B., Birkholtz L.M., Joubert F., Haider N., Walter R.D. and Louw A.I. (2007). Structural and mechanistic insights into the action of Plasmodium falciparum spermidine synthase. Bioorg. Med. Chem. 15, 1628-1637. [ Links ]
56. de Beer T.A.P., Louw A.I. and Joubert F. (2006). Elucidation of sulfadoxine resistance with structural models of the bifunctional Plasmodium falciparum dihydropterin pyrophosphokinase-dihydropteroate synthase. Bioorg. Med. Chem. 14, 4433-4443. [ Links ]
57. Tastan Bishop A.O., Wells G., Joubert F., Wrenger C., Walter R.D. and Louw A.I. (2007). Progress towards the structure of Plasmodium falciparum pyridoxal kinase. In Proceedings of First Southern African Bioinformatics Workshop, pp. 41-44. [ Links ]
58. Sewell B.T., Thuku R.N., Zhang X. and Benedik M.J. (2005). Oligomeric structure of nitrilases effect of mutating interfacial residues on activity. Ann. N.Y. Acad. Sci. 1056, 153-159. [ Links ]
59. Sewell B.T., Berman M. and Meyers P.R. (2003). The cyanide degrading nitrilase from Pseudomonas stutzeri AK61 is a two-fold symmetry, 14-subunit spiral. Structure 11, 1413-1422. [ Links ]
60. Jandhyala D., Berman M.N. and Meyers P.R. (2003). CynD, the cyanide dihydratase from Bacillus pumilus: gene cloning, and structural studies. Appl. Environ. Microbiol. 69, 4794-4805. [ Links ]
61. Thuku N.R., Weber B.W., Varsani A. and Sewell T. (2007). Post-translational cleavage of recombinantly expressed nitrilase from Rhodococcus rhodochrous J1 yields a stable, active helical form. FEBS J. 274, 2099-2108. [ Links ]
62. Cameron R.A., Sayed M. and Cowan D.A. (2005). Molecular analysis of the nitrile catabolism operon of the thermophile Bacillus pallidus RAPc8. Biochim. Biophys. Acta 1725, 35-46. [ Links ]
63. Karl-Eric J. and Eggert T. (2002). Lipases for biotechnology. Curr. Opin. Biotechnol. 13, 390-397. [ Links ]
64. Litthauer D., Ginster A. and van Eeden Skein E. (2002). Pseudomonas luteola lipase: a new member of the 320-residue Pseudomonas lipase family. Enzy. Microbiol. Technol. 30, 209-215. [ Links ]
Author for correspondence. E-mail: ozlem@tuks.co.za

