










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.103 n.9-10 Pretoria Sep./Oct. 2007
REVIEW ARTICLE
A framework for the informed normalization of printed microarrays
Johan van Heerden; Sally-Ann Walford; Arthur Shen; Nicola Illing
Department of Molecular and Cell Biology, University of Cape Town, Private Bag, Rondebosch 7701, South Africa
ABSTRACT
Microarray technology has become an essential part of contemporary molecular biological research. An aspect central to any microarray experiment is that of normalization, a form of data processing directed at removing technical noise while preserving biological meaning, thereby allowing for more accurate interpretations of data. The statistics underlying many normalization methods can appear overwhelming to microarray newcomers, a situation which is further compounded by a lack of accessible, non-statistical descriptions of common approaches to normalization. Normalization strategies significantly affect the analytical outcome of a microarray experiment, and consequently it is important that the statistical assumptions underlying normalization algorithms are understood and met before researchers embark upon the processing of raw microarray data. Many of these assumptions pertain only to whole-genome arrays, and are not valid for custom or directed microarrays. A thorough diagnostic evaluation of the nature and extent to which technical noise affects individual arrays is paramount to the success of any chosen normalization strategy. Here we suggest an approach to normalization based on extensive stepwise exploration and diagnostic assessment of data prior to, and after, normalization. Common data visualization and diagnostic approaches are highlighted, followed by descriptions of popular normalization methods, and the underlying assumptions they are based on, within the context of removing general technical artefacts associated with microarray data.
Introduction
DNA microarrays allow for the large-scale quantitation of gene expression by inference of mRNA transcript abundance.1 Since its inception, the technology has developed to become an essential item in the biologist's arsenal of tools. Microarray-based techniques rely heavily on various statistical methods for the preparation and analysis of the high-throughput data generated in these experiments. The large numbers, and nature, of variables associated with microarray experiments require novel statistical procedures. These methods present a new challenge to the molecular biologist, requiring a paradigm shift from classic one-gene-at-a-time approaches, to techniques that evaluate thousands of genes simultaneously. An aspect central to any microarray experiment is that of normalization, a form of data processing directed at removing technical noise or systematic variation while preserving biological meaning, thereby allowing for more accurate interpretations of data.2
In recent years, biologists have been confronted with a multitude of publications detailing purportedly new and advanced algorithms for the normalization of microarray data. The effectiveness of many algorithms, at reducing error, has been evaluated by using data sets of which sample ratios are known a priori.3,4 Prompted by these studies, we provide an introductory review on the performance and robustness of several commonly used algorithms, highlighting the assumptions that these methods are based on and suggesting an approach to normalization that could be useful when encountering microarray-based techniques for the first time. An ad hoc approach is encouraged, recognizing that each microarray experiment will have unique requirements, which have to be identified before deciding on a normalization strategy.
The need for normalization
Microarray experiments allow biologists to investigate gene expression patterns of thousands of genes in a single assay. The observed patterns of expression in any microarray experiment are affected by several sources of variation, which can obscure true biological values and impede meaningful interpretations.5 Variation can be divided into two broad categories: (1) the interesting kind, which has biological meaning and is of value to the researcher; (2) the obscuring kind, also referred to as noise or systematic variation, which has no meaning and is a result of the technical error rather than the experimental design.6 The aim of normalization is to account for these artefactual contributions while preserving the true biological meaning of the observed expression values.
Sources of experimental noiseª have been well documented,5,7 and considering their effect on microarray data is an important aspect of any normalization strategy. Systematic errors can be introduced at various points during a microarray experiment, from sample preparation to hybridization and scanning. These errors appear as inconsistencies in the generated data, which can be identified by various diagnostic and visualization tools.5 Failure to correctly identify and correct for systematic error can lead to results becoming obscured to the point of not containing any biological meaning.8 There are many alternative methods of normalization available to a researcher. Deciding which normalization algorithms to apply to their data, and being able to substantiate a chosen strategy, are among the challenges faced by researchers.
Experimental design, normalization and the role of controls
It is pertinent to note that printed microarrays commonly come in one of two flavours, referred to as single-or dual-channel arrays. With the former, a single biological sample is labelled and hybridized to an array surface; the latter involves two independent biological samples, each labelled with different fluorophores. This distinction is important, as certain types of technical artefacts occur only in dual-channel arrays. Where necessary, these differences will be highlighted in the text.
When approaching normalization strategies, it is important to realize that underlying many of the algorithms are certain assumptions about the nature of data being normalized. Commonly used normalization algorithms often assume two things: (1) that the majority of genes in a microarray experiment are not differentially regulated, i.e. remain unchanged, and (2) that the number of up-regulated genes are more or less equal to the number of down-regulated genes.2,8,9 While these assumptions might be accurate for arrays that include all or most of the genes in a genome, it cannot be assumed to be valid for arrays that include only a subset of genes, often referred to as custom or directed arrays.5 When the above assumptions are invalid, control spots will be essential to any chosen normalization strategy, where they can be used as stable references for the validation and normalization of microarray data.8
Careful experimental design, at the outset of an experiment, cannot be overemphasized. Researchers should, a priori, consider possible normalization strategies based on the content of their array slides. It is important that there is no bias in the types of gene targets printed in different sections of the slide. No normalization strategy will be reliable if the appropriate controls are not included as part of the design. The choice of control spots will differ, depending on the type of array platform and the facility at which the array is produced; common approaches include the use of synthetic spots, housekeeping genes or the identification of a set of genes that are known to be invariant or unresponsive across conditions assayed. All these approaches aim to provide some kind of calibration reference, i.e. a set of spots for which expression values can be predicted beforehand, which can be used to validate and normalize microarray data. Any deviations from expected or predicted values can be considered a result of systematic bias. A bias factor can then be calculated using the control spots and its effect extrapolated to the rest of the spots on the array.5,8
For control spots to be effective, they should (1) span the entire intensity range, (2) be distributed randomly across the surface of an array, and (3) be numerous enough to provide a statistically robust reference.8 It is desirable to include, as part of the design of an experiment, flexibility with regard to several possible normalization options.
Data diagnostics and visualization
Identifying systematic bias via data exploration
A first step, in deciding upon the most appropriate method of normalization, is to visualize the patterns of variation in the raw data.b Identifying the nature of technical interference allows the researcher to decide on a directed normalization strategy, one that preserves biological data while reducing the noise specific to the array data set. Approaching normalization blindly, without assessing raw data, introduces a real danger of silencing or removing biological information of interest.8,10 This is as detrimental to the outcome of an experiment as the non-removal of technical noise.
Systematic errors, which have discrete local effects on subsets of data on an array (e.g. all the spots printed by a specific pin), are called local biases. In contrast to this, systematic errors which have a general or global effect across an entire data set are referred to as global biases. Accordingly, normalization algorithms address the contribution of systematic errors either globally or locally.2 The implication of this is that global normalization methods assume a general smooth error trend across a data set, while local methods assume that the source of bias affects discrete subsets of data independently from other such discrete units. The scope of normalization chosen—global or local—should be dictated by, and complement, the nature of a specific bias.8 Using diagnostic and visualization methods to explore data, allows the researcher to determine whether technical noise or systematic errors produce within-and/or between-slide variations, and whether these biases exhibit global or local behaviours.
Several diagnostic and visualization tools exist and are available in most microarray analysis software packages. Most common and useful among these are: 1) box plots, 2) histograms, 3) scatter and MA-plots, and 4) false-colour plots. Each method allows for the identification of specific traits of the data and facilitates an evaluation of the contribution of unwanted noise. Interpretation of visualizations depends on experimental design and requires careful consideration. For directed or custom arrays, it is imperative that all diagnostic interpretations are substantiated by control spotsc or known invariant genes, as any predictions or assumptions regarding the distribution of feature values can be problematic.
Box plots5
Box plots are commonly used to assess the relative spread of data, usually log ratiosd or feature intensities, and are therefore a convenient way of identifying scale differences within or between arrays (Fig. 1). These plots provide a graphical overview of the so-called five-number summary of a data set, which includes information about the three quartiles [i.e. 25th percentile, 50th percentile (also called the mean), and the 75th percentile] and the minimum and maximum values. This tool can be used to compare the spread of data points from different print-tips, different microtitre plates or the overall spread of data from different arrays.
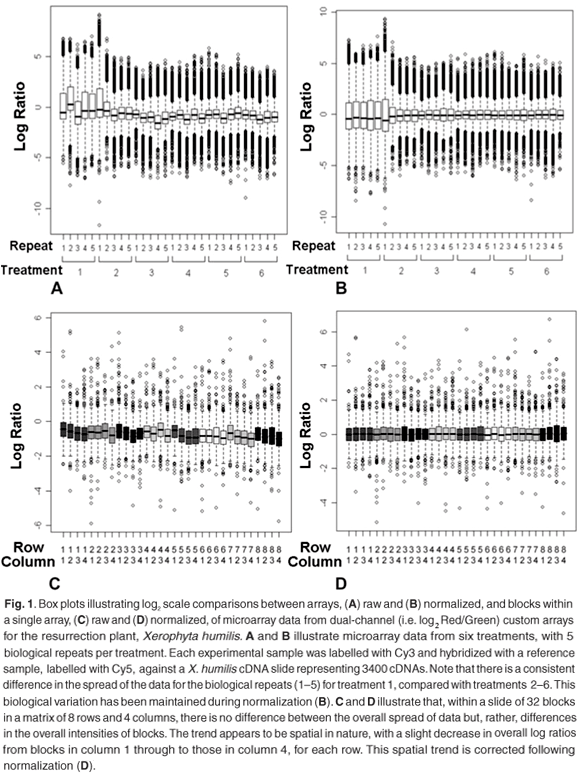
Overall scale differences between blocks can be the result of inconsistencies between pins or the non-random distribution of genes during printing. The latter can be eliminated by good experimental design. Assuming that experimental design has allowed for an unbiased distribution of gene targets, scale differences can result from variations in the amount of target deposited by different print-tips, which results in differences in the relative brightness of blocks of spots.10 A second source of variation that could contribute to scale differences within cDNA arrays is that of microtitre plates. Different cDNA amplification batches are usually associated with different microtitre plates; inconsistent conditions show up as variations in spot intensities between replicate spots picked from different batches.11
Any differences in the spread of data between arrays could be a result of differences in scanner settings used to scan each array,12 i.e. photomultiplier tube (PMT) and laser voltage settings, differences in mRNA concentrations isolated from samples, or differences in the labelling conditions of samples. Care should be taken when interpreting scale differences between arrays, as these differences could also reflect real experimental conditions which, if corrected for, will introduce rather than remove noise.11
Figure 1 shows an example of treatment-induced scale differences, where these differences reflect biological responses, rather than technical inconsistencies. Control spots can be a useful calibration guide for making sure that scanner settings are set correctly at the point of data capture of fluorescent signals from custom slides, to reduce between-slide scale differences.
Identifying scale differences and their probable sources will allow the researcher to adjust correctly for these. Emphasis is again directed at informed interpretation of the observed behaviour. Correcting for scale differences between print-tipse or microtitre plates, when these differences are a result of the non-random distribution of genes, will do nothing but introduce noise and silence biological meaning. Similarly, between-array scale differences could be condition-or treatment-specific and should be judiciously evaluated.
Histograms5
It is often useful to visualize information regarding the shape of the distribution of generated data. Histograms are plots of the frequency of feature intensity values or log ratios. Information regarding distributional density, i.e. number of values and their relative occurrence, across a data set, can be gleaned from these plots. Such information is useful when comparing the equivalence of distributions between two data sets. Some between-array normalization algorithms assume the data between arrays to be equally distributed.8 Histograms also provide information regarding the central tendencies and absolute values of data sets, similar to box plots. In addition, these plots are useful when trying to ascertain whether the given data are normally distributed, as this is a requirement for many parametric statistical analysis techniques to be valid. The visualization of intensity distributions from custom arrays is particularly important, where assumptions regarding the distributional nature of data can be problematic.
Scatter plots5,11
Scatter plots provide a useful means for comparing the behaviour of different dyes in dual-channel experiments or, alternatively, comparing the relative overall behaviour between arrays (single-or dual-channel arrays). When comparing two arrays, the ratio values of features from each array are plotted on the x-and y-axis, respectively. If two arrays behave similarly, that is, the overall log ratios or feature intensities of individual features are comparable, then the points within such a plot will approximate a straight line with a slope of one and an intercept of zero. When comparing replicate arrays (or control spots from non-replicate arrays), any deviation from the expected straight line is indicative of systematic error.
More commonly, the same logic is used to compare the behaviour of two different dyes (Fig. 2A). Any deviations from a straight line with slope one and intercept zero is indicative of systematic differences between the two dyes. A useful type of scatter plot is the so-called MA-plotf (also referred to as the ratio-intensity, or RI, plot), which is used to identify inconsistencies or biases in the behaviour of two different dyes, across the entire feature intensity range, in dual-channel experiments. The MA-plot is essentially a normal scatter plot, of which the axis has been shifted by 45° and then scaled (Fig. 2B). The average log intensity {A = ½[log(Ch1i) + log(Ch2i)]} of features is plotted against the log2 ratio [M = log(Ch1i) – log(Ch2i)] of these features, yielding a horizontal axis around which points are distributed. The x-axis values (average intensity, or A) can be calculated as log2 or log10. For large random data sets, the assumption is that points should be distributed more or less symmetrically around a log ratio of zero. This assumption should carefully be considered when working with small or custom arrays. It is not uncommon to see a tailing of values at extreme intensity ranges, often referred to as the 'banana-effect'. This type of artefact can be ascribed to differences in the fluorescent capacities, or quantum yields, at different intensities, and differential incorporation of the dyes, due to differences in the size of Cy3 and Cy5 molecules.5,8,12
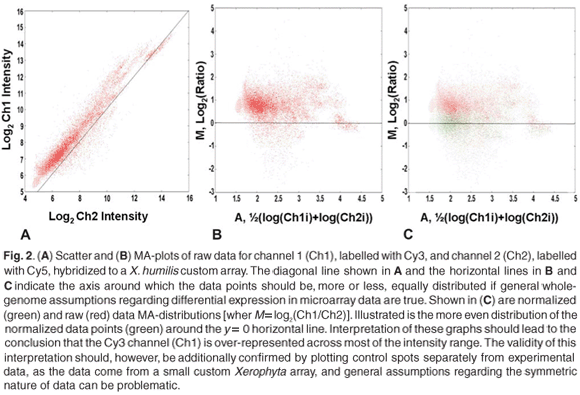
Identifying this type of bias is clearly important if some kind of reliable comparison, between samples labelled with different dyes, is to be made. When comparing the behaviour of dyes, the MA-plot has an important advantage over a normal scatter plot: points are plotted along a horizontal axis rather than a diagonal one – the human eye and brain are more efficient at interpreting horizontal distributions than diagonal ones11 (Fig. 2B).
False-colour plots8,11
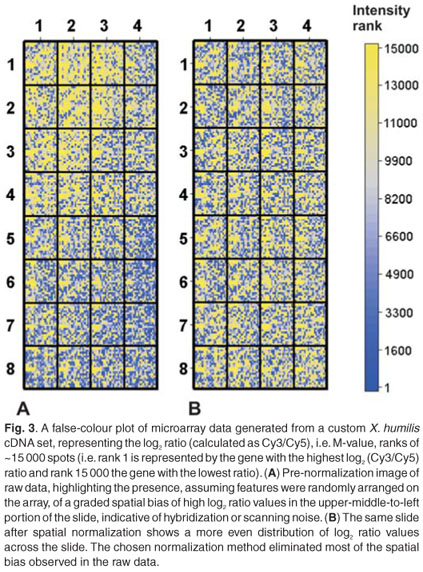
These kinds of plots are commonly used to identify spatial bias, which has been found to affect many arrays. Spatial bias refers to the effect that a specific feature's two-dimensional position has on its intensity value. False-colour plots can be generated by plotting the log transformed ratio or intensity value of a feature, as a function of its xy-coordinates in an array, or alternatively as a rank value, again as a function of its xy-coordinates. Spatial trends can easily be identified in this kind of plot and can be seen as a non-random distribution of log transformed ratios or intensity values. This type of bias can be introduced as a result of differences between microtitre plates or print-tips, hybridization artefacts, inserting slide into scanner at an angle, imperfections on the glass slide or any other effects related to the optical properties of microarray technology. Differences between print-tips lead to a specific type of spatial bias, where discrete blocks of features appear to be distinctly different from other blocks. Differences between microtitre plates lead to a similar discrete pattern of spatial bias. Imperfections on the glass slide and scanner-based variables can cause either discrete or smooth spatial effects (Fig. 3). It is important to identify and correctly interpret the behaviour of spatial bias. Choosing a normalization algorithm that corrects for discrete spatial effects, when these effects are smooth and global, will introduce noise. Similarly, a global spatial correction method should be chosen if the bias does not exhibit discrete behaviour and feature distribution is known to be random. Special care should be taken when interpreting false-colour plot data from small custom arrays, where clusters of differentially regulated genes might appear as spatial artefacts. A global normalization method will silence the biologically meaningful information contained within these small, differentially regulated clusters.
Measures of replicate variability
The visualization tools outlined above form a central part of pre-analysis data exploration, assisting the researcher in identifying the nature and extent to which data are affected by systematic error or technical noise. As illustrated above, these visualization tools are equally useful for the post-normalization state evaluation of data, providing some insight regarding the efficacy of a chosen method at removing noise. The visualization and subsequent interpretation of normalized data, however, should be approached cautiously. Applying inappropriate normalizations to data can potentially introduce noise which might not necessarily show up as an increase in noise when using the visualization tools. Logic, however, dictates that replicate features within arrays, or between biological repeats, should exhibit equivalent behaviour, with any deviations being indicative of the effects of unwanted systematic error or technical noise. The extent to which normalization minimizes or reduces variation across replicate features can therefore be used as a reliable means of assessing the efficacy of any normalization strategy in addition to the visualization methods already noted. Estimations of variability for replicate features are commonly obtained using pooled variances or ANOVA models, amongst others. It is assumed that the better a normalization method addresses the specific biases present in a data set, the smaller the variation among replicated observations will become.12
The issue of backgroundg
More than any other bias factor, background contributions and their bias effect have been hotly debated in the literature. Although background subtraction is not the focus of this review, a brief consideration of its effect on data and normalization is warranted. Background refers to the contribution to overall spot intensity by targets binding non-specifically to the support matrix as well as fluorescence by the glass slide itself;11 it is therefore reasoned that this leads to an over-estimation of target abundance for specific features. It has been shown that methods aimed at removing this bias often introduce, rather than remove, noise. In addition, the choice of array platform can greatly affect the performance of background subtraction and its effect on overall noise reduction.4 Khojasteh et al.4 noted significant differences in the efficacy of background subtraction, when applied to copy number (CGH) data generated by SMRT (Sub Mega base Resolution Tiling) array and cDNA array platforms, respectively. Data from the SMRT arrays showed a higher degree of reliability when background values were subtracted, whereas the cDNA array data showed the opposite behaviour. Khojasteh et al.4 ascribed the apparent need to subtract background values from SMRT array-derived data to platform specific behaviour in addition to the specific image analysis methods used for these types of arrays.
Background bias can be measured globally or locally. Global measures assume a general linear trend or contribution across the array, while local approaches assume a more discrete contribution. Commonly used methods for the estimation of a background bias factor involve: (1) the inclusion of unrelated gene sequences or the inclusion of 'blank' spots on the slides, which are used to estimate global background fluorescence, or (2) the estimation of local background fluorescence in the area immediately surrounding a spot.5 These approaches all aim to arrive at a reliable estimation of a non-specific contribution to foreground fluorescence. The intention of background subtraction is a valid one, but its effectiveness is debatable. First, it is questionable whether contributions made by non-specific hybridization are significant enough to warrant correction. Second, assuming that background contributions are non-trivial, local estimations of background by image analysis software are based on the incorrect assumption that the relative contribution of background to overall fluorescence is linear. This assumption is not valid, as the relative contribution of background to the overall intensity of spots within the high intensity range will be much less than the contribution experienced by spots in the lower intensity range. Third, methods attempting to determine the non-specific interaction effect between targets and unrelated gene sequences are erroneous in that background contribution is based on chemical interactions between the glass matrix and nucleic acids, the chemistry of which is profoundly different from that observed in interactions between unrelated nucleic acid species.5 Lastly, methods based on estimations of local background are notoriously unreliable, with the choice of image analysis software having a major effect on the outcome of estimation.4,13 Many background subtraction algorithms that aim to account for the non-linearity of local contributions are available. These algorithms modify calculated background values before subtracting them from calculated feature values. These modifications typically attempt to avoid common logical paradoxes, such as negative feature values, that arise from linear estimations and simple subtractions of background.
The effect background subtraction has on the efficiency of subsequent normalization depends very much on the accurate identification and removal of a true background bias factor. Special care should be taken not to confuse background with spatial effect, which is better corrected for by spatial normalization methods.8 False-colour plots, discussed above, can also be used to explore the nature of background bias by plotting estimated background values against xy-coordinates.10 Incorrectly estimating background contribution will do nothing but increase noise, which is clearly undesirable.3,14 Situations resulting in negative values for features are not uncommon8 and are indicative of the shortcomings of the specific methods. Wit and Mclure8 propose some approaches to background subtracting that aim to avoid this logical dilemma. It is advisable, in addition to visualizing the background contribution, to assess diagnostically the effect of background subtraction on data, once applied.
The literature does not provide a clear-cut answer to the issue of background subtraction, and although its application might be theoretically warranted, it is important to consider whether attempts at estimating background bias are robust enough to be reliable. A fairly simple global strategy is highlighted by Wit and McClure.8 The method uses empty spots as measures of the lowest achievable value on the array and then estimating a background bias factor based on the average intensity of these control spots. This central tendency, preferably median, can be subtracted from overall intensity values of features, setting any resulting negative values to zero. This method, however, requires that empty spots be sufficiently numerous and that their distribution cover a reasonable representation of the array surface, for a reliable background bias factor to be estimated. In addition, it is based on the assumption that the background bias is constant across the array. A consensus regarding the need for, and reliability of, background subtraction is yet to be reached; recent publications are, however, progressively leaning towards a 'no background subtraction' approach.4,8
Normalization methods
Numerous normalization algorithms exist, each one designed to correct for specific systematic errors introduced during a microarray experiment. This section will consider normalization methods in the context of the type of technical variation they address. Many of these methods are equally applicable to one-and two-colour arrays; where this is not the case, it will be highlighted. This discussion does not aim to provide a definitive list of normalization strategies, but rather attempts to highlight the bias factors being addressed by each method and some assumptions upon which these algorithms are based.
A recommendation regarding a generally reliable and proven strategy is made, following the discussion of normalization methods. This recommendation is based on studies involving the empirical validation of various normalization methods. Empirical studies conducted by Qin et al.3 and Khojasteh et al.4 have provided a wealth of useful information regarding the performance of the algorithms on data sets with known expression values.
A case-by-case interactive approach is encouraged, which should involve the empirical exploration of data, via the diagnostic and visualization tools highlighted above, before and after normalization. Once the specific biases, affecting an experiment, have been identified, a stepwise normalization framework can be compiled. Emphasis is again directed at the two assumptions underlying many normalization methods: (1) that the majority of genes in a microarray experiment are not differentially regulated (they remain unchanged), and (2) that the number of up-regulated and down-regulated genes is more or less equal.2,9 In addition, some between-array normalization algorithms also assume a similar distribution of expression values, that is, the frequency of specific intensity values (or log ratios) is approximately equivalent between arrays.8 These methods are referred to as parametric, which means that some kind of explicit assumption regarding the distribution of the data is made, this is in contrast to non-parametric methods.5 Whenever these assumptions are not satisfied, a set of invariant genes, or control spots, for which true values can be predicted should be used to determine bias factors to be extrapolated to the rest of the data set. This effectively allows the researcher to use empirically validated parametric methods to predict a bias effect on non-parametric data. Most normalization algorithms, where applicable, allow the researcher explicitly to define control spots or a set of invariant genes to be used.
Colour correction/Dye bias
Intensity bias, also referred to as intensity-dependent dye bias, is a result of slight differences in the properties of the commonly used Cy3 and Cy5 dyes. Using scatter plots, mentioned in the Data diagnostics and visualization section above, highlights these kinds of biases clearly. The effect is often seen as a 'tail' of spots in the lower and/or higher intensity ranges, indicating inconsistent behaviour of dyes.8 This type of bias affects only dual-channel arrays, so the methods considered here will be discussed in this context. There are several different algorithms which address and correct for this type of bias.
1. Linear regression
Simple linear regression was one of the first methods used in early microarray experiments but is generally no longer used. These techniques are included to provide some historical perspective.
a. Dye vs dye
This method involves the adjustment of one channel (e.g. Cy3 vs Cy5) relative to the other (Fig. 2A), based on the distributional assumptions regarding microarray data symmetry. A best-fit line is generated through the distribution of spots on a scatter plot of Cy3 vs Cy5 (or Cy5 vs Cy3) values. The gradient and intercept of this best-fit line is then calculated, with deviations from a slope of one, and a y-intercept of zero, being taken as a reflection >of inherent noise. The linear equation therefore provides a normalization function that can be used to adjust values such that the slope is equal to one with a y-axis intercept equal to zero. This is simply done by adjusting all x-axis values with the 'deviating' slope and intercept values (i.e. xnorm = mx + c). Note, however, that this type of linear regression treats Cy3 and Cy5 channels differently, which is not desirable as the assignment of dyes to different axes produces different results.11
b. MA
Linear regression-based normalization can alternatively be done on data distributed within an MA-plot (Fig. 2B). A best-fit line is again calculated through the points in the plot. Normalized M-values are calculated by subtracting the fitted value, for each feature, from the raw log ratio (i.e. M-value). Using MA-plotbased linear regression has the advantage that the two channels are treated equally, which makes these regressions more robust and reproducible.11
Neither of these methods is recommended for correcting the intensity-dependent bias often observed in dual-channel arrays. It has been noted that the intensity-dependent effect is non-linear, with the implication that a linear model will not be able to account effectively for this kind of bias.11
2. Lo(w)ess based methods
Lo(w)ess (LOcally WEighted Scatterplot Smoothing, or LOcally WEighted polynomial regreSSion) is a type of non-linear regression commonly used to adjust the distribution of points in an MA-plot. All Lo(w)ess algorithms employ the same strategy for the modelling of bias and subsequent adjustment of values. Adjustments are made, as with linear regression, by subtracting fitted values from raw log ratios. This method performs a series of local regressions across the MA-plot, using sliding windows of predefined size.16 These local regressions are then combined into a single smooth curve.5,11 The non-linear fits generated by Lo(w)ess algorithms are able to account more reliably for intensity-dependent dye bias than linear methods. The distributional assumptions highlighted earlier pertain to standard Lo(w)ess algorithms, as these employ a weighting function for determining the centre of a collection of data points. Lo(w)ess algorithms try to fit data around an M-value (log2 ratio) of zero. Points further away from this assumed mean are deemed more unreliable than those closer to it and therefore contribute less to the position of a centroid within a collection of points.2
Various parameters control the behaviour of a Lo(w)ess adjustment. First, a Lo(w)ess regression can be performed using, in theory, polynomials of any degree, which affects the nature of the generated best-fit line. It is common practice to fit data around polynomials of degree one, i.e. a straight line, as it has been observed that higher-degree polynomials (e.g. binomial, trinomial, etc.) tend to over-fit data, which does not capture the general trend in a population of data points.5 The assumption is that within a certain range, the intensity-dependent bias is linear, and using a series of sliding windows will ensure the fitting of spots within a linear range. The size of the sliding window, the second parameter, influences the reliability and sensitivity of the Lo(w)ess algorithm and typically determines the proportion of points to include in each regression calculation.5 Setting this window size too small again results in over-fitting, while setting this window too big results in a Lo(w)ess curve that does not model, effectively, the non-linear nature of the bias. The Lo(w)ess regression windows overlap, hence the designation sliding window, with the result being a very large number of overlapping regression calculations. Lo(w)ess >algorithms can be computationally demanding for this reason, but this is becoming less of a problem with powerful desktop computers. It should be noted that Lo(w)ess algorithms can be sensitive to outliers, despite the weighting approach mentioned earlier. Where data points are limiting, the statistical robustness,
reliability, of each regression can become unreliable in the presence of large numbers of outliers due to an increase in the relative contribution made by each data point.
a. Global Lo(w)ess
This approach uses all data points within a given array to generate a non-linear curve, which is used to adjust for intensity-dependent dye bias.4,8 Global Lo(w)ess generally performs quite well at estimating a dye bias, provided the observed bias is not a result of other systematic errors (e.g. underlying spatial bias).8
b. Composite Lo(w)ess10
This type of Lo(w)ess provides a slightly more advanced global method for dye bias adjustment. Concerns regarding the reliability of Global Lo(w)ess adjustments within the extreme intensity ranges have been raised, as these ranges typically contain fewer data points than intermediate intensity ranges. Composite Lo(w)ess is based on a model where control spots as well as assayed features are used to generate a non-linear best-fit line. The idea is that as the sliding window moves into extreme intensity ranges, the Lo(w)ess curve will be increasingly based on the control spots rather than the assayed features, which will contribute increasingly fewer data points to the window. Because this type of Lo(w)ess relies on both assayed features and control spots, it is not a viable normalization method for data sets that rely on control spots only for the calculation of a bias factor.
c. Print-tip Lo(w)ess
In contrast to the above global methods, Print-tip, also known as block-by-block, Lo(w)ess employs a discrete local strategy for the modelling and correction of dye bias. Print-tip Lo(w)ess is used to adjust feature intensity values printed by each pin separately. The principle remains the same, but the assumptions are slightly different. Print-tip Lo(w)ess assumes that each discrete block of features will behave slightly different from other blocks due to minor physical differences between pins. Print-tip Lo(w)ess can simultaneously correct for intensity-dependent and spatial bias (discussed below).10 Concerns regarding the discrete nature of this type of Lo(w)ess approach should be noted. There is a danger of introducing bias in cases were there is no discernible difference in the overall intensity-dependent behaviour of features from different blocks.8 It has also been noted that Print-tip Lo(w)ess is unreliable in cases where there are fewer than 150 data points per print-tip group. In these cases, a global method provides a statistically more robust approach as the number of data points used to model the bias is substantially more.10
3. Dye-swap normalization8
Another method commonly used to account for differences in the fluorescent capacity of the different dyes is that of dye-swap normalization. This method relies on the inclusion of technical replicates as part of an experiment's design. Typically, an array will be replicated with the samples in each replicate, labelled with opposite dyes. The intensities of the replicate features are then calculated as average intensities across both dyes.
This method can be extended to include Lo(w)ess fitting, once average intensities are calculated for replicate features, thereby providing a very robust model for intensity-dependent correction. Including dye-swap replicates, as part of a dual-channel experiment, is theoretically highly desirable, but often practically unfeasible, due to the high cost of microarray experiments.
4. Splines17,h
Spline-based algorithms present a non-parametric alternative to the Lo(w)ess-based regression fitting of data, and are commonly used to account for intensity-dependent dye bias. The main benefit of spline-based normalization methods is their independence from the assumptions underlying other parametric approaches. Spline algorithms do not make any assumptions regarding the distributional nature of data, but rather treat values simply as a collection of points and are therefore useful when normalizing directed or custom arrays.
This method is related to Lo(w)ess in that it is based on the calculation of several local regressions, across a data set, which are joined to form a smooth curve. Spline-based methods, however, are based on a discrete window approach, as opposed to the overlapping sliding window method employed by Lo(w)ess algorithms. Spline algorithms perform a fixed number of linear regressions, within a predefined number of windows, across a data set. Spline-based dye bias normalizations are usually implemented in a manner equivalent to the Lo(w)ess methods discussed above, using MA-distributions. The behaviour of a spline curve can be modified via parameters similar to those used to modify Lo(w)ess regressions. Typically, the polynomial degree of the curve is specified; as with Lo(w)ess, it has been found that higher-degree polynomials often over-fit data, with linear equations generally performing best. In addition, the number of windows, i.e. number of regression calculations, across a data set is defined. This is distinctly different from Lo(w)ess methods, where the number of regression calculations for any predefined window size can differ, depending on the number of data points within a data set. Spline-based algorithms are computationally much less intensive than Lo(w)ess algorithms, due to the usually significantly smaller number of regression calculations performed.
The various methods discussed above can be extended to include several robust parameters, which add dimensions to data sets. One such extension, suggested by Smyth and Speed,10 involves ranking the quality of spots and assigning a reliability weight to features when applying Lo(w)ess regressions to the data. Rank Weighted Lo(w)ess involves calculating a centroid of a collection of data points based on the statistical reliability of features. Reliability is measured as a percentage of the complement of pixels that make up each spot. Spots with more pixels are deemed to be more reliable than those with fewer and consequently carry more weight in determining the centroid of a specific collection of data points.
Spatial bias
This type of bias, if present, can clearly be seen in the false-colour plots discussed earlier.11 Unlike intensity-dependent bias, this type of systematic variation affects both single-and dual-channel arrays.
1. Lo(w)ess based methods
Again, Lo(w)ess based algorithms can be used to model the spatial effect observed on many arrays. The assumptions and parameters are the same as previously stated.
a. Print-tip Lo(w)ess
As noted above, Print-tip Lo(w)ess can be used to correct simultaneously for intensity-dependent bias as well as spatial bias. The assumption is that spatial trends are localized to discrete areas of the array and can therefore be accounted for by adjusting values within discrete units, in this case print-tip groups or blocks.10 The same curve used to correct for intensity-dependent bias is used to adjust spatial trends within a print-tip group, with adjustments based on MA-distribution regressions. Because of the discrete nature of this approach, there is a danger of introducing noise at the edges of a print-tip group when the underlying spatial effect is continuous across the surface of an array.8 It is therefore important to determine whether the spatial trends observed in false-colour plots are discrete or continuous before applying Print-tip Lo(w)ess. Another important consideration is the number of features associated with each print-tip. As mentioned previously, Print-tip Lo(w)ess is unreliable in cases where there are fewer than 150 data points per print-tip group.10
b. 2D Lo(w)ess
This type of spatial correction is effective for the removal of continuous spatial trends. Regression fitting of values is based on trends seen within two-dimensional false-colour plots. As with other Lo(w)ess methods, polynomial curves are used to model non-linear trends within data. Wit and McClure8 recommend using Lo(w)ess polynomials of degree one, i.e. linear functions, when correcting for spatial trends, as it has been observed that higher-degree polynomials tend to be unstable near the edges of microarrays. 2D Lo(w)ess assumes a global spatial trend which, as mentioned above, might or might not be the case. Again, an assessment of spatial trends is necessary before making any adjustments. 2D Lo(w)ess might not be the best option in cases where imperfections on the array present sudden rather than smooth changes, or in cases where clusters of differentially expressed genes are found. 2D Lo(w)ess will confuse such clusters with spatial bias that has to be adjusted for.11 These aspects require consideration by the researcher and care should be taken to avoid any unbiased distribution of gene targets during printing at the outset of the experiment.
2. Median based methods4
An alternative to the Lo(w)ess methods discussed above is a spatial correction method based on the central tendency of neighbourhoods of spots. For each spot, the median of log2 intensity values of spots within a spatial neighbourhood of predefined size (number of rows × number of columns), centred on that spot, is calculated. The difference between the neighbourhood median and the intensity value of the spot is considered to be a bias factor. The value of each spot is adjusted accordingly. The neighbourhood size used to adjust the value of spots is an important parameter to consider. A small neighbourhood is sensitive and corrects discrete and local artefacts, but might be problematic when adjusting for more general or global trends (compare Global vs Print-tip Lo(w)ess). A large neighbourhood size will clearly have the opposite effect. The choice of neighbourhood size depends on diagnostic interpretations of the spatial bias effect.
Scale differences
Scale biases are common to both single-and dual-channel arrays and methods correcting for this kind of systematic error are generally applicable to both platforms. All scaling methods have two things in common: (1) they adjust the means of compared data sets to be more or less equal (also known as centring) and/or (2) adjust the spread or variation of data to be more similar (also known as scaling). Data sets can consist of any collection of measurements, for example, values associated with specific microtitre plates or print-tips, values associated with control spots, and values associated with one or multiple arrays.
1. Subtract log2 central tendency
This method adjusts the means of all distributions to zero. It is one of the simplest forms of scale adjustment and involves subtracting either the log2 mean or median of a distribution from each feature's log2 ratio. This results in the mean of all distributions, adjusted in this way, being equal to zero. This method can also be applied to the raw ratios; in this case, all ratios are divided by the measure of central tendency (i.e. mean or median).11 This technique works well, but ignores possible array-wide changes which might be a reflection of sample conditions or treatment. It is therefore advisable to approach global scaling methods with caution. As with all normalization methods, controls can be used to calculate an adjustment factor. Another objection, concerning this method, involves assumptions regarding the linearity of variation. Bright arrays exhibit compression of values at high intensities, whereas darker arrays show compression of values near low intensities. This behaviour is a consequence of the limit imposed on possible intensities values for any feature (0 to 216 –1), which lead to a breakdown in linearity at the extremes of the intensity range.8
2. Subtract log2 central tendency and divide by standard deviation
This form of scaling adds another dimension to that of the method discussed above. In addition to adjusting the means of all distributions to zero, the standard deviations of these distributions are brought to one. The same comments and considerations discussed above apply to this method.
3. Quantile normalization
Quantile normalization was proposed as a method for the scaling of replicate arrays, where assumptions based on whole-genome expression distributions are problematic, but works equally well on whole-genome arrays. This method forces the distribution of values in each array in a set of arrays to be the same. All features are ranked according to their intensity value— that is, the lowest intensity value is assigned rank 1; the second-lowest intensity value is assigned rank 2, etc.—until all features within each slide have been ranked. The ranked distributions are then compared and the mean of each rank across the arrays is calculated. This calculated mean replaces the original value and the normalized data are rearranged to have the original ordering.8 This type of approach ensures an equivalent distribution of intensity values between distributions. Wit and McClure8 point out that this method is able to deal with the non-linear compressions that might affect the two scaling approaches mentioned above, as the ranking approach ensures a linear distribution of features.
When adjusting the scale of replicate slides with this method, it is reasonable to assume that the distribution of feature intensities should be comparable. This assumption, however, is often problematic, as different slides usually do not have the same number of captured features. This is commonly a result of technical thresholds or noise (e.g. detection of low signals or washing artefacts) and less often biological differences.8 A potential problem is that slides with different numbers of features have a different number of ranks. Features with missing values across arrays should therefore be excluded or imputed, prior to attempting quantile normalization. Several methods for the imputationi of missing values exist,18,19 a detailed discussion of which falls outside the scope of this review. Some implementations of the quantile algorithm20 overcome the problem of missing values by assuming that missing values are random and not a result of low signal or technical noise. By implication, this means that the number of missing values should be proportional to the number of features on an array. Missing values that result from non-random effects (commonly due to a low signal) are therefore still problematic as these invalidate the assumption of the quantile algorithm (G.K. Smyth, pers. comm.). As previously emphasized, stated assumptions are often erroneous and should be validated before proceeding with any type of normalization. Histograms provide a useful way for visualizing and comparing the density distributions of different data sets.
One approach to between-condition or -treatment scaling is based on the ranking of a set of controls or invariant genes, i.e. genes that are known not to change between conditions. A general ranked scale is then generated and the remaining genes are linearly distributed, known as interpolation, between the ranks of the invariant genes. This approach can also be used when there are large differences in the number of data points within distributions. For this method to be reliable, the smallest and largest values on each array have to be part of the set of invariant genes.8
Dual-channel arrays, which are based on a common reference design, offer an intuitively interesting solution to between-slide scale adjustments. Sample-or treatment-channels may not show a comparable distribution of intensity values across the different experimental conditions. If use is made of a common reference, however, this by definition should have the same distribution across all slides, and channel-specific implementations of quantile adjustments20 can be considered. These methods essentially force the reference channel across all slides to be exactly the same and extrapolate an adjustment factor for the sample-or treatment-channel within each array.
4. Cyclic Lo(w)ess
This is an inter-array variant of the previously mentioned Lo(w)ess-based methods originally developed for cDNA microarrays. This algorithm can be applied to both single-and dual-colour arrays to adjust for scale differences between them. An MA-plot is generated, where M is defined as the log2 ratio of replicate feature values and A as the average of replicate feature log values.6,13 Generating an MA-plot in this way allows for a comparison of features across replicate arrays. As with other Lo(w)ess methods, a non-linear regression curve is calculated and the data are fitted accordingly. This process is carried out in a pair-wise manner and iterated until differences between arrays have been removed. This procedure can be applied to sets of invariant genes or control spots in cases where the features on arrays are not expected to be directly comparable.13 The same Lo(w)ess parameters, previously discussed, are applicable to this specific implementation of the Lo(w)ess algorithm.
5. Qspline
This is referred to as a baseline method and, similar to Cyclic Lo(w)ess, is an inter-array variant of its intra-array counterpart. Estimations and adjustments are dependent on the definition of a baseline array, also called a reference array. The baseline array is used as a ranking reference for subsequent adjustments. Target array features are ranked and compared to the ranked features of the baseline array. A spline-based smoothing curve (discussed above) is then calculated to relate the ranks of features from the target array to those on the baseline array.6 The choice of baseline array is important and can have a profound effect on results. The baseline array should ideally be representative of an average behaviour of replicate features across the different arrays. This essentially means that the feature values on the baseline array should preferably be more or less equal to the mean value of those features across all arrays. It is not necessary that the baseline array contain all features; it can be constructed using a set of known invariant genes which occur across all arrays, or other forms of control spots for which expression values can be expected to be similar. Quantile ranks are then constructed using these genes, with the resultant smoothing function being extrapolated to other features of an array.13
ANOVA-based methods
ANOVA-based methods of normalization have been shown to be effective in modelling systematic error. These methods do technically more than just normalize data; they also provide estimations regarding the significance of condition-specific gene expression, a feature which falls outside the scope of this discussion. This method is based on a composite linear model, which contains terms for each aspect of the array and all possible sources of bias. Interactions between the different aspects of an array (e.g. genes, dyes, print-tips, spatial position, and time point) and the specific sources of bias or error are then mathematically defined. Variations between features are assessed in the context of various null hypotheses and their significance statistically determined. The null hypotheses usually state that the observed variation is not significant but a product of systematic bias. If these hypotheses are rejected, the observed variation is presumed to be biologically meaningful. In this way, ANOVA methods distinguish between interesting and obscuring variations.5 The problem with such an approach is the non-linear nature of many artefacts associated with systematic error. An ANOVA-based model is not able to estimate these non-linear artefacts, a good example of which is the non-linear intensity-dependent dye effect described earlier.5,8
Evaluation of normalization methods
A comprehensive study, conducted by Khojasteh et al.,4 showed that a composite stepwise approach to normalization provides the most reliable means of identifying and removing the systematic errors. They compared the efficacy of different combinations of normalization models at detecting single-copy gene changes between samples, for which the gene copy ratios were known. In total, 19 different normalization strategies were assessed across five different data sets, all with different numbers of arrays. Performance was based on a specific strategy's ability to reduce variability (standard deviation) and enhance the accuracy of predicting single-copy gene changes between samples. Normalization strategies consisted of single-step (addressing only one type of bias), two-step (addressing two sources of bias), and three-step (addressing three sources of bias) approaches. The types of biases addressed were: (1) spatial bias, (2) intensity bias, and (3) global scale bias. All normalization strategies were performed including and excluding background subtraction. The results were unanimous, indicating that a three-step strategy, one that systematically addresses the three most common sources of variation, without background subtraction, outperformed both the two-and one-step approaches. Two-step strategies, in turn, outperformed the one-step approaches, highlighting the importance of identifying and correcting for all sources of systematic error.
Conclusion
The sources of systematic variation that affect a microarray experiment are many and accounting accurately for each of them is not trivial. Although a variety of normalization strategies have been developed to identify and correct for these systematic biases, these strategies are based on stringent assumptions which require careful consideration. The conscientious design of a microarray experiment, the inclusion of appropriate controls and the unbiased printing of gene targets are imperative to the successful normalization of microarray data.
We recommend a stepwise strategy that systemically addresses the various types of biases identified with the diagnostic and visualization tools discussed earlier. It should be emphasized that a thorough diagnostic interpretation of data, prior to normalization, facilitates the compilation of a normalization strategy aimed at addressing directly the types of systematic error present within a specific experiment. The effectiveness of each normalization strategy should be diagnostically monitored before proceeding with the next step. Wit and McClure8 suggest that local artefacts should be corrected before progressing to normalizations that involve several or all arrays. These recommendations are empirically supported by the results of Khojasteh et al.4 In particular, Wit and McClure suggest the following systematic strategy:
1. Spatial correction
2. Background correction
3. Intensity-dependent dye bias
4. Within-replicate scaling
5. Between-condition/-treatment scaling.
Wit and McClure8 point out that, although background subtraction is included in the above list, they advise strongly against its use. This advice is supported by the study of Khojasteh et al.4
Many other more complex algorithms exist, a list too long to include here, but their reliability and performance remain questionable due to insufficient empirical validation. Many of these purportedly novel algorithms are derivations of commonly used ones and essentially address the same types of biases. A new trend towards non-parametric algorithms—algorithms that make no explicit assumption regarding the distributional nature of the data—can be seen in the literature. Whether these methods provide a real advantage over current parametric models depends on results obtained from empirical validations, where a priori statements regarding expression values can be made.
Recommended resources
Many commercial and open source solutions are available to microarray researchers. Choosing among the various options is often a matter of personal preference, as most of the available software packages, aimed at microarray data analysis, contain a large selection of visualization and diagnostic tools. A good starting point is one of several microarray portals (some listed below), which include descriptions of software packages as well as links to other useful resources.
1. http://www.biodirectory.com/biowiki/Microarray_portals_and_resource_pages
A site that is used by life-science researchers to find tools and databases related to all sorts of molecular biological and bioinformatics-related activity, including experimental troubleshooting, tutorials, as well as applications and methods-related information.
2. http://www.microarrays.in/links.html
A great one-stop resource for microarray software, protocols, links, publications and other microarray-related information including discussion forums.
3. http://cbio.uct.ac.za/arrayportal
A portal containing introductory information on important microarray-related topics, including links to useful references.
We thank Wiesner Vos, from the UCT National Bioinformatics Node, for many useful discussions and advice on normalizing data from custom microarrays. This work was supported financially by the University of Cape Town and the National Research Foundation. J. v. H. thanks Dan Stein, Department of Psychiatry, University of Cape Town, for financial support during his Honours year in the Department of Molecular and Cell Biology.
Notes (refer to superscripts in text)
a) Experimental noise comes in many forms and is often referred to as technical, or systematic, error, variation or noise. In this paper these terms are all used to refer to the unwanted, biologically meaningless, variability observed within, or between, arrays.
b) Technical variation can exist within an array, that is, between features printed on the same slide, or between arrays, i.e. between the features from different slides.
c) Spots and features are both terms used to refer to the genes present on an array. In addition, each spot or feature can be considered a discrete collection of data, or data point.
d) It is common for intensity values to be log-scaled. Most often this is done as log2. Advantages of the log-scaling of data include the linearization and symmetrization of the distribution of feature intensities. In addition, log2-scaling makes data more amenable to subsequent interpretation.5
e) Print-tips refer to the pins used by the spotting robot to spot DNA sample onto the array. Commonly, each pin is associated with a discrete group, also called a block of spots.
f) It is important to consider the way in which expression ratios are represented, when interpreting MA-plots. In MA-plots ratios are commonly calculated as Cy5/Cy3, but can equally be done as Cy3/Cy5.
g) Background estimation and subtraction refer to two different procedures. Estimation refers to the process of calculating raw background values whereas subtraction refers to the method by which estimated values are processed.
h) Traditionally, a spline is defined as a long, narrow, flexible strip of timber. The definition of a mathematical spline is analogous to its timber counterpart and refers to a flexible mathematical function able to adapt to data.17
i) Imputation refers to the process by which values for features with missing values are derived from patterns inherent in existing data.
1. Schena M., Shalon D., Davis R. and Brown P.O. (1995). Quantitative monitoring of gene expression patterns with a cDNA microarray. Science 270, 467–470. [ Links ]
2. Quackenbush J. (2002). Microarray data normalization and transformation. Nature Genetics Suppl. 32, 496–501. [ Links ]
3. Qin L. and Kerr F. (2004). Empirical evaluation of data transformations and ranking statistics for microarray analysis. Nucleic Acids Res. 32, 5471–5479. [ Links ]
4. Khojasteh M., Lam W.L., Ward R.K. and MacAulay C. (2005). A stepwise framework for the normalization of array CGH data. BMC Bioinformatics 6, 274. [ Links ]
5. Drãghici S. (2003). Data Analysis Tools for Microarrays. Chapman & Hall/CRC Press, London. [ Links ]
6. Bolstad B.M., Irizarry R.A., Astrand M. and Speed T.P. (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193. [ Links ]
7. Hartemink A., Gifford D., Jaakkola T. and Young R. (2001). Maximum likelihood estimation of optimal scaling factors for expression array normalization. SPIE International Symposium on Biomedical Optics 2001 (BiOS01). In Microarrays: Optical Technologies and Informatics, eds M. Bittner, Y. Chen, A. Dorsel and E. Dougherty, Proc. SPIE, 4266, 132–140. [ Links ]
8. Wit E. and McClure J. (2004). Statistics for Microarrays: Design, Analysis and Inference. John Wiley, Chichester, Hants. [ Links ]
9. Wang D., Huang J., Xie H., Manzella L. and Soares M.B. (2005). A robust two-way semi-linear model for normalization of cDNA microarray data. BMC Bioinformatics 6, 14. [ Links ]
10. Smyth G.K. and Speed T.P. (2003). Normalization of cDNA microarray data. Methods 31(4), 265–273. [ Links ]
11. Stekel D. (2003). Microarray Bioinformatics. Cambridge University Press, Cambridge. [ Links ]
12. Park T., Yi S., Lee S., Lee Y. and Simon R. (2003). Evaluation of normalization methods for microarray data. BMC Bioinformatics 4, 33. [ Links ]
13. Wu W., Dave N., Tseng G.C., Richards T., Xing E.P. and Kaminski N. (2005). Comparison of normalization methods for Codelink Bioarray data. BMC Bioinformatics 6, 309. [ Links ]
14. Wu W., Xing E.P., Myers C., Mian I.S. and Bissell J.M. (2005). Evaluation of normalization methods for cDNA microarray data by k-NN classification. BMC Bioinformatics 6, 191. [ Links ]
15. Sen A. and Srivastava M. (1990). Regression Analysis: Theory, Methods, and Applications. Springer-Verlag, New York. [ Links ]
16. Cleveland W.S. and Devlin S.J. (1988). Locally weighted regression: An approach to regression analysis by local fitting. J. Am. Stat. Assoc. 83, 596–610. [ Links ]
17. Rupert D., Wand M.P. and Carrol R.J. (2003). Semiparametric Regression. Cambridge University Press, Cambridge. [ Links ]
18. Sehgal M.S.B., Gondal I. and Dooley L.S. (2005). Collateral missing value imputation: a new robust missing value estimation algorithm for microarray data. Bioinformatics 21, 2417–2423. [ Links ]
19. Acuna E. and Rodriguez C. (2004). The treatment of missing values and its effect in the classifier accuracy. In Classification, Clustering and Data Mining Applications, eds D. Banks et al., pp. 639–648. Springer-Verlag, Berlin, Heidelberg. [ Links ]
20. Smyth G.K. (2005). Limma: linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor, eds R. Gentleman, V. Carey, S. Dudoit, R. Irizarry and W. Huber, pp. 397–420. Springer, New York. [ Links ]
Received 16 April. Accepted 1 September 2007.
Author for correspondence. E-mail: nicola.illing@uct.ac.za

