










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkOnderstepoort Journal of Veterinary Research
On-line version ISSN 2219-0635
Print version ISSN 0030-2465
Onderstepoort j. vet. res. vol.81 n.2 Pretoria Feb. 2014
PROCEEDINGS
Molecular survey for foot-and-mouth disease virus in livestock in Tanzania, 2008-2013
Raphael S. SalluI, II; Christopher J. KasangaII; Mkama MathiasI, II; Mmeta YongoloI; Chanasa Mpelumbe-NgelejaI; Misheck MulumbaIII; Ezekia RangaIV; Philemon WamburaII; Mark RweyemamuII; Nick KnowlesV; Donald KingV
ITanzania Veterinary Laboratory Agency, Dar es Salaam, Tanzania
IISouthern African Centre for Infectious Diseases Surveillance, Sokoine University of Agriculture, Tanzania
IIISouthern African Development Community Secretariat, Gaborone, Botswana
IVMinistry of Livestock Development and Fisheries, Dar es Salaam, Tanzania
VWorld Reference Laboratory for Foot-and-Mouth Disease, Institute for Animal Health, United Kingdom
ABSTRACT
Phylogeography data are of paramount importance in studying the molecular epidemiology dynamics of foot-and-mouth disease virus (FMDV). In this study, epithelial samples and oesophageal-pharyngeal fluids were collected from 361 convalescent animals (cattle and buffaloes) in the field throughout Tanzania between 2009 and 2013. The single plex real-time RT-PCR (qRT-PCR) assay for rapid and accurate diagnosis of FMDV employing the Callahan 3DF-2, 3DF-R primers and Callahan 3DP-1 probe were used. Preparation of the samples was performed according to the OIE manual, with a Kenya O serotype obtained from the attenuated vaccine serving as a positive control and samples collected from healthy animals serving as true negatives. The results indicated that 53.49% of samples (n = 176) were positive for FMDV genome by qRT-PCR, with Ct values ranging from 14 to 32. In addition, molecular typing of the FMDV genome positive samples using serotype specific primers revealed the existence of several serotypes: serotype South Africa Territory 1 (SAT1) (34.25%, n = 60), serotype A (68.92%, n = 98), serotype O (59.20%, n = 98) and SAT2 (54.54%, n = 96). The virus protein 1 sequences analysis for 35 samples was performed and the collective results indicated: 54.28% serotype O, 25.71% serotype A, 14.28% serotype SAT1 and 2.85% serotype SAT2. Therefore in this study, both the phylogenetic trees and spatial distribution of serotypes elucidated the phylodynamics of multiple FMDV field strains in Tanzania and neighbouring countries.
Introduction
Foot-and-mouth disease virus (FMDV) belongs to the genus Aphthovirus, family Picornaviridae which principally infects cloven-hoofed animals including wildlife animals. The virus is a positive sense, single-stranded RNA virus and is categorised into seven serotypes: A, O, C, Asia 1, South Africa Territory 1, 2 and 3 (SAT1, SAT2 and SAT3) (Domingo et al. 2003). FMDV can be genetically classified based on its geographic origin (topotypes); for example, the serotype SAT1 can be grouped into eight topotypes (I-VIII) based on nucleotide differences (within virus protein 1 [VP1] coding sequence) of up to 15% (Samuel & Knowles 2001). The serotype SAT1 topotype III is found in Tanzania, Zambia, Malawi, Kenya and Zimbabwe according to the study conducted by Vosloo et al. (2002).
Foot-and-mouth disease (FMD) is highly contagious and, combined with its high antigenic diversity, this makes the disease difficult to control. The disease has caused significant economic losses as a result of the deaths of young animals, decreased productivity and trade sanctions against livestock and animal products from infected regions. This disease is endemic in most countries of sub-Saharan Africa (Vosloo et al. 2002) and is commonly reported in Southern African Development Community (SADC) countries. To date, efforts to control this disease have not been very successful. This is contributed mainly to the lack of knowledge on the actual FMDV serotypes and subtypes (topotypes) circulating in these countries. The knowledge of the serotypes and subtypes is a prerequisite for effective regional disease control strategies.
The epidemiology of FMD in Tanzania is influenced by two different patterns, viz. a cycle in which wildlife plays a role in maintaining and spreading the disease to other susceptible domestic animals and wild ungulates and a cycle that is maintained within domestic animals and that is independent of wildlife (Kivaria 2003). In southern Africa, the former cycle predominates as a result of the presence of the African buffalo (Syncerus caffer), the only wildlife species for which long-term maintenance of FMDV has been described (Hedger, Forman & Woodford 1973). In the SADC region, both cycles probably occur, whilst in West Africa, because of the absence of significant numbers of wildlife hosts, the virus is believed to be maintained primarily within the domestic animal cycle. In southern Africa, a number of countries have been able to control FMDV by separating infected buffalo from livestock and by limited use of vaccination.
From the epidemiological surveillance conducted by Rweyemamu et al. (2008), it has been observed that Tanzania lies in the Great Lakes cluster, which comprises the countries of the East African Community (i.e. Tanzania, Kenya, Uganda, Rwanda and Burundi), as well as the eastern part of the Democratic Republic of Congo (DRC). This region not only has large livestock populations but also the highest concentration of wildlife in the world. Farming is dominated by agro-pastoral communities and is characterised by communal grazing and migrations. The eastern DRC is heavily dependent on trade livestock from Uganda, Tanzania, Rwanda and Burundi. This Great Lakes cluster probably contains the most complicated FMD situation in the world, with several FMD primary endemic foci. Five serotypes (O, A, C, SAT1 and SAT2) are endemic in this cluster and the sixth serotype (SAT3) was isolated in wildlife (African Buffalo) in Uganda in 1970 (Hedger et al. 1973); although, it has never been isolated from the livestock in the Great Lakes cluster. In Tanzania, the common bovine FMDV serotypes observed are SAT1, SAT2 and O. These serotypes were identified by C-ELISA Antigen during a period of seven years, 2002-2009. The serotypes were observed from the following regions, namely: Kagera (type O), Kilimanjaro (SAT2), Morogoro (SAT2), Pwani (SAT1 and SAT2), Rukwa (SAT2), Singida (SAT2) and Tabora (SAT1) (Central Veterinary Laboratory 2009). The previous effort to control this disease showed futile results, as there are reported cases where there was a re-emergence of the disease after the animals have been vaccinated. Also, the use of quarantine and culling strategies are very expensive to execute and difficult because of the geography of the country.
Research method and design
Viruses
The viruses used in this study were obtained from all endemic areas of Tanzania from the outbreak cases from 2008 to 2013. Collection of the samples followed the World Organisation of Animal Health (OIE) guidelines (OIE 2012) and included all bio-data such as the location where the animal was found, name of the area and farmer, type of animal, age and sex. Besides this, also the type of the farming system practiced by farmer was noted, that is zero grazing or range. The epithelium, probang and serum samples were collected from the clinical animals, especially cattle and pigs, which normally show disease symptoms. The epithelium samples were taken from the affected muzzle, gums, tongue and hooves. The oro-pharyngeal samples were stored in liquid nitrogen, whilst the epithelium samples were stored in a 50:50 mixture of PBS and glycerol at pH of 7.2 and stored in cold chain as serum samples. Before the epithelium tissues were ground, drying was performed using the blotted dry absorbent to reduce glycerol content and weighed. An epithelial suspension was prepared by grinding the sample in sterile pestle and mortal with sterile sand. The suspension was centrifuged at 2000 rpm for 10 min and the supernatant taken for the molecular work.
RNA extraction
The RNA extraction was conducted directly from the 361 samples from epithelium tissues suspension, sera and oro-pharyngeal sample using QIAamp® Viral RNA kit (Qiagen, Hilden, Germany). Besides the 361 samples, extraction was also conducted to additional samples to form a positive extracted control (serotype O Kenya vaccine strain) and a negative extracted control.
The 361 samples were tested using real RT-PCR as described by Shaw et al. (2007). Samples were screened using the Callahan 3D probe (6-FAM 5' - TCC TTT gCA CgC CgT ggg AC - 3' TAMRA), 3D forward (5' - ACT ggg TTT TAC AAA CCT gTg A - 3') and 3D reverse (5' - gCg AgT CCT gCC ACg gA - 3') primers (Eurogentec, Liege, Belgium). The master mix kit used in this assay was InVitrogen Superscript III Platinum One Step qRT-PCR System (Cat. No. 11732-020), with the final volume per reaction as 20.0 µL, plus 5.0 µL from each template RNA. The reaction mix per one reaction was as follows: 2x reaction mix at 12.5 µL, nuclease free water 1.0 µL, primer 3DF 2.0 µL (10 µM), primer 3DR 2.0 µ! (10 µM), probe 3DP 1.5 µL (10 µM), ROX 1:10 pre-diluted Superscript III RT 0.5 µL and Platinum Taqmix 0.5 µL.
The PCR regime was as follows: 30 min for 60 °C (reverse transcriptase step), 10 min at 95 °C (inactivation reverse transcriptase or activation DNA polymerase), 15 s for 95 °C (denaturation) and 1 min at 60 °C (annealing and elongation), with the two last steps performed for 50 cycles. Measurement fluorescence was taken at the end of second step at stage 3. The results interpretation was as follows: samples with Ct < 32 were classified as positive, samples with 32 < Ct < 50 were ambiguous and marked for retesting and samples with no Ct were classified as negative. The cycle threshold value (Ct value) was fixed automatically from the pre-loaded machine software (7500 software v. 2.0.5). FMDV positive samples were further analysed for RT-PCR and VP1 sequencing.
Real-time PCR and VP1 sequencing
One primer combination was used for the RT-PCR of FMDV serotypes, as follows: serotype O: O-1C244F/EUR-2B52R, serotype A: A-1C612F/EUR-2B52R, serotype C: C-1C536F/ EUR-2B52R, serotype Asia1: As1-1C530F/EUR-2B52R, serotype SAT1: SAT1-1C559F/SAT-2B208R, serotype SAT2: SAT2-P1-1223F/SAT-2B208R and serotype SAT3: SAT3-P1-1222F/SAT-2B208R (Table 1). One-step RT-PCR was carried out using the Qiagen One-step RT-PCR kit (Cat. no. 210210).
The thermal profiles used for amplification of the VP1 sequence of various serotypes were as follows: FMDV O and Asia1: 42 °C for 30 min, 94 °C for 5 min, 35 cycles at 94 °C for 60 s, 60 °C for 60 s and 72 °C for 90 s, followed by a final extension of 72 °C for 5 min. Conditions were the same for the other serotypes, except that extension temperatures were 55 °C for A and C and 50 °C for SAT1, SAT2 and SAT3. Temperature cycling was carried out using the GeneAmp® PCR systems 9700 (Applied Biosystems, Foster City, USA).
Standard 2% (w/v) agarose gel (SeaKem® LE Agarose, Rockland, USA) was prepared by dissolving 3 g agarose in 150 mL 1x TBE electrophoresis buffer (0.040 M Tris-Borate, 0.001 M EDTA, pH 8.0) in an Erlenmeyer flask. The mixture was heated in a hot plate or microwave to allow the agarose to dissolve and form a gel. The gel was allowed to cool to about 20 °C before adding 3 µL of ethidium bromide (10 mg/mL). The gel was then poured into a horizontal gel tray fitted with appropriate combs. After about 40 min of gel polymerisation, the combs were carefully removed and the tray immersed in an electrophoresis tank containing electrophoresis buffer (1x TBE). Then 3 µL of DNA products of each individual sample was mixed with 2 µL of bromophenol blue dye diluted 6x (3:1 water: 6x dye, 50 Mm EDTA, 50 mM NaCl, 50% glycerol) and then loaded into separate lanes (slots) of the submersed agarose gel. The samples were run at 120 volts for 45 min. After the run, the gel was removed and photographed under UV light using a video capture system (Molecular Imager® Gel Dox XR System 170-8170 with Flowgen IS 1000; Bio-Rad, Seoul, Korea).
Sequences and phylogenetic analyses
The PCR products were directly sequenced on both strands to obtain the complete VP1 sequences, which were compared with the other relevant FMDV VP1 sequences within the same serotypes. Computer-assisted comparisons of the nucleotide sequences were made to find the similarities of nucleotides sequences in the National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), using the BLASTN search program.
Also, for further sequence comparisons, another sequence resource used was from the European Bioinformatics Institute (http://www.ebi.ac.uk/service/) maintained by the European Molecular Biology Laboratories. VP1 sequences data were assembled and analysed using SeqMan II (DNAStar Lasergene 8.0, Madison, USA). This led to final sequence of 636-639 nucleotides of the VP1 coding region. Nucleotide sequences that encoded amino acid from the VP1 gene were translated to the (deduced) amino acid sequences by the EditSeq. To compare with ingroup sequences, additional sequences from the GenBank were used for the VP1 region. Alignment of VP1 sequences was performed using Clustal W algorithm method (MegAlign; DNAstar, Madison, USA) and Bio-Edit 7.0. These alignments were used to construct distance matrices by using the Kimura 2-parameter nucleotide substitution model in the program MEGA 5.1 (Tamura et al. 2007). Midpoint-rooted neighbour- joining trees were then constructed with MEGA 5.1 software. The robustness of the tree topology was assessed with 1000 bootstrap replicates by using the model in MEGA 5.1.
Results
Real-time PCR and typing
The results from different primer combinations (Table 1) indicated that 53.49% of samples (n = 176) were positive for FMDV genome by qRT-PCR, with Ct values ranging from 14 to 32. In addition, molecular typing of the FMDV genome positive samples using serotype specific primers revealed the existence of several serotypes: serotype SAT 1 (34.25%, n = 60), serotype A (68.92%, n = 98), serotype O (59.20%, n = 98) and SAT 2 (54.54%, n = 96). The VP1 sequences analysis collectively across all 35 samples was performed and the results indicated: 54.28% serotype O, 25.71% serotype A, 14.28% serotype SAT1 and 2.85% serotype SAT2.
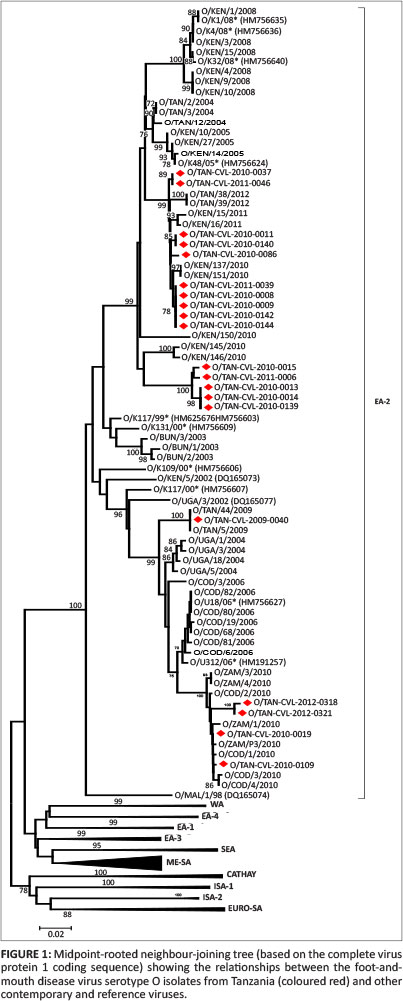
Phylogenetic analysis of serotype O, A and SAT1 viruses
Complete VP1 sequence analysis data from the analysed samples, together with the archived data from across Africa, were used. Serotype O predominated the FMDVs examined, with all 20 of the FMDV serotype O Tanzania isolates genotyped in this study clustered into a single topotype: East Africa (EA)-2 (Figure 1). The results from different parts of Tanzania revealed that the same topotype is circulating and is responsible for causing the majority of outbreaks. Isolate O/TAN-CVL-2010-0037 is 100% similar to O/TAN-CVL-2011-0046, which is from the Tabora region. The phylogenetic analysis from these isolates concurs with the geographical distribution of the virus with respect to outbreaks (Kasanga et al. 2012). The serotype O viruses were found in Tabora, Serengeti, Musoma, Kahama, Mwanza, Rorya, Iringa, Mbeya, Rukwa and the Pwani region, which amounts to the majority of the country.
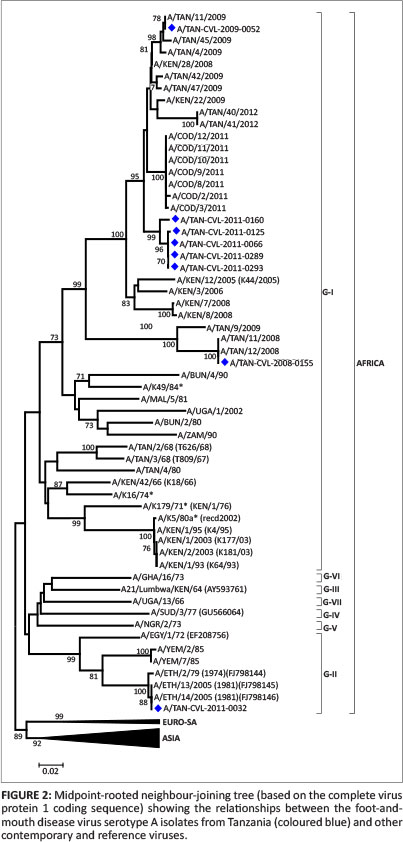
All the isolates in this serotype were grouped in the AFRICA topotype (Genotype 1) of the serotype viruses (Figure 2) as it was previously identified in the country (Kasanga et al. 2012). The isolates A/TAN/11/2008 and A/TAN/12/2008, which were reported to be 100% similar, are also similar to isolate A/TAN-CVL-2008-0155 from the Rukwa region, with high bootstrap support. Isolate A/TAN/11/2009 was 100% similar to isolate A/TAN-CVL-2009-52 from Tabora. For the serotype A topotype AFRICA (G1), the recorded nucleotide difference in VP1 coding region of 0.0% - 15.6%, with any other topotypes defined for FMD Type A viruses reported in Africa. These serotype A viruses were found in regions such as Mtwara, Tabora, Kigoma, Rukwa and Dar es Salaam and this shows the virus is expanding its territory, as previously reported (Kasanga et al. 2012).
For the phylogenetic analysis of SAT1 (Figure 3), it can be concluded that all FMD type SAT1 viruses isolated in Tanzania and analysed to date were placed within the topotype 1NWZ and the nucleotide differences in VP1 coding region of 15% - 60%. The nucleotide number of composition was 663 and the average nucleotide match was from 645-660 with the unknown strain. All bootstrap of 70% and above are shown (Figure 3). The isolates numbered SAT1/TAN-CVL-2012-0359 and SAT1/TAN-CVL-2012-0361 are 100% similar and were obtained in the same area in the Morogoro region. The five SAT1 isolates used in this study, SAT1/TAN-CVL-2012-0354, SAT1/TAN-CVL-2012-0355, SAT1/TAN-CVL-2012-0359, SAT1/TAN-CVL-2012-0360 and SAT1/ TAN-CVL-2012-0361 are clustered together in a group with isolates SAT1/TAN/11/2012, SAT1/TAN/23/2012, SAT1/ TAN/25/2012 and SAT1/TAN/27/2012 obtained from the northern part of Tanzania (Figure 3). Since the detection of SAT1 in the country in 1971 (Rweyemamu & Loretu 1972, 1973), this serotype has been responsible for many outbreaks, especially in the wildlife and livestock interface.

Discussion
This study on the molecular survey of FMDV was focused on the viruses circulating in Tanzania from 2008 to 2013. The phylogenetic analysis of VP1 sequences has been widely used to identify and characterise FMDV isolates (Klein et al. 2006; Knowles et al. 2009; Konig et al. 2007). The final study included 18 type O, 9 type A and 6 SAT1 field isolates collected from different endemic areas of country. The results showed that the O type is the one responsible for most of the outbreaks in the country. The serotype O is endemic and has spread widely in the country since the late 1950s (Rweyemamu 1972; Vosloo et al. 2002). This serotype seems to be widespread in the northern, southern, western and eastern regions of Tanzania, but not in the central parts of the country (Kasanga et al. 2012). Serotype O, together with the A and SAT1 serotypes, are the primary causes of sporadic emergencies in some parts of Tanzania (Kasanga et al. 2012).
The previous studies indicated some enigma relating to the dynamics of Serotype A in Tanzania. But from 2009 to 2012, the incidence dynamics of this serotype started to change; in 2009 there was one outbreak in Nzega (Tabora) caused by isolate A/TAN-CVL-2009-0052. This isolate is 100% similar to the one reported by Kasanga et al. (2012), which caused the outbreak in the eastern coastal zone. But, in this current study, it was revealed that the serotype A is spreading fast in Tanzania, as it was found in Nzega, Mtwara, Tabora, Kibondo (Kigoma), Dar es Salaam and Sumbawanga (Rukwa). In a previous study (Kasanga et al. 2012), this serotype was only reported in regions such as Iringa, Morogoro, Pwani, Dar es Salaam and Dodoma. According to Rweyemamu and Loretu (1972), Serotype A was regularly reported in the northern, northern-lake and central zones of Tanzania up to 1971. The samples collected and analysed from 1997 to 2004 revealed no presence of the serotype A (Swai, Mrosso & Masambu 2009). With this current study, it seems that this serotype's reappearance is contrary to the previous reports. The spread and reappearance of serotype A could possibly be caused by the movement of animals across the border or within the country by nomadic livestock keepers. Therefore, a detailed study should be conducted regarding to the disappearance and reappearance of serotype A, which makes a genetic comparison between the old and the new strains which are appearing.
Conclusion
The present study provides a comprehensive picture of the phylogenetic relationships of FMDV isolates, focusing on the circulating viruses in Tanzania, thus filling a gap of knowledge in the country regarding the molecular epidemiology of this economically important virus. As FMDV is rapidly spreading in the country, further collection and analysis of samples, together with the improvement of the local epidemiological investigation of FMD outbreaks is recommended. Intensive phylogeography studies should be carried out that will locate particular serotype or topotype that is responsible for the disease outbreak. This is a very important step in controlling the spread of the disease and also for the production of a specific vaccine which will cater for a specific topotype. In conclusion, this study provides an overview of the molecular epidemiology of FMDV in Tanzania. Although only a limited number of ad hoc samples were available for the sequence analysis, this study highlights the spread of FMDV in Tanzania.
Acknowledgements
The authors thank the researchers and staff from the Veterinary Investigation Centres for sample collection during FMD outbreaks. We also appreciate the assistance provided by farmers during the surveillance exercises. We recognise the contribution of staff at the Centre for Infectious Disease and Biotechnology - Temeke during the laboratory analysis of samples. We are grateful to the World Reference Laboratory, Pirbright, UK which assisted us during the data analysis. The Ministry of Livestock Development and Fisheries, United Republic of Tanzania funded the technical advice. The authors also acknowledge the Welcome Trust Foundation through Southern African Centre for Infectious Disease Surveillance (SACIDS) for providing financial support for PhD work. Last, but not least, the first author wishes to thank all his supervisors at Sokoine University of Agriculture, World Reference Laboratory for Foot and Mouth Disease-Pirbright, Tanzania Veterinary Laboratory Agency and the Royal Veterinary College. This work was supported by the Wellcome Trust Grant WT087546MA to the Southern African Centre for Infectious Diseases & Surveillance (SACIDS).
Competing interests
The authors declare that they have no financial or personal relationships that may have inappropriately influenced them in writing this article.
Authors' contributions
R.S.S. (Tanzania Veterinary Laboratory Agency) was involved in the laboratory work, surveillance and data analysis. P.W. (Sokoine University of Agriculture), M.R. (Sokoine University of Agriculture) and C.J.K. (Sokoine University of Agriculture) were the first authors' supervisors in laboratory work and academic advisors. M.Y. (Tanzania Veterinary Laboratory Agency), M.M. (Tanzania Veterinary Laboratory Agency) and C.M.-N. (Tanzania Veterinary Laboratory Agency) where the first authors' technical advisors during surveillance work and sample preparation work. D.K. (World Reference Laboratory for the Foot and Mouth Disease-Pirbright) and N.K. (World Reference Laboratory for the Foot and Mouth Disease-Pirbright) were involved in data analysis.
References
Central Veterinary Laboratory, 2009, Virology annual report, Ministry of Livestock and Fisheries Development, Centre for Infectious Diseases, Temeke, Dar Es Salaam-Tanzania. [ Links ]
Domingo, E., Escarmis C., Baronowski, E., Ruiz-Jarabo, C.M., Carrillo E., Neun~z. J.I. et al., 2003, 'Evolution of foot-and-mouth disease virus', Virus Research 91, 47-63. http://dx.doi.org/10.1016/S0168-1702(02)00259-9 [ Links ]
Hedger, R.S., Forman A.J. & Woodford M.H., 1973, 'Foot-and-mouth disease in East African buffalo', Bulletin of Epizootic Diseases of Africa 21, 99-101. [ Links ]
Kasanga, C.J., Sallu, R., Kivaria, F., Mkama, M., Masambu, J., Yongolo, M. et al., 2012, 'Foot-and-mouth disease virus serotypes detected in Tanzania from 2003 to 2010: Conjectured status and future prospects', Onderstepoort Journal of Veterinary Research 79(2), Art. #462. http://dx.doi.org/10.4102/ojvr.v79i2.462 [ Links ]
Kivaria, F.M., 2003, 'Foot-and-mouth disease in Tanzania: An overview of its national status', Veterinary Quarterly 25, 72-78. http://dx.doi.org/10.1080/01652176.2003.9695147 [ Links ]
Klein, J., Parlak, U., Ozyoruk, F. & Christensen, L.S., 2006, 'The molecular epidemiology of foot-and-mouth Disease virus serotype A and O from 1998 to 2004 in Turkey', BMC Veterinary Research 2(35), 1-13. [ Links ]
Knowles, N.J., Nazem Shirazi, M.H., Wadsworth, J., Swabey, K.G., Stirling, J.M., Statham, R.J. et al., 2009, 'Recent spread of a new strain (A-Irn-05) of foot-and-mouth disease virus type A in the Middle East', Transboundary Emerging Diseases 56, 157-169. http://dx.doi.org/10.1111/j.1865-1682.2009.01074.x [ Links ]
Konig, G.A., Palma, E.L., Maradei, E. & Piccone, M.E., 2007, 'Molecular epidemiology of Foot-and-Mouth disease virus types A and O isolated in Argentina during the 2000-2002 epizootics', Veterinary Microbiology 124, 1-15. http://dx.doi.org/10.1016/j.vetmic.2007.03.015 [ Links ]
OIE World Organisation for Animal Health, 2012, Terrestrial animal health code. Chapter 8.5: Foot-and-mouth disease, OIE, Paris. [ Links ]
Rweyemamu, M.M. & Loretu, K., 1972, 'Observations on foot-and-mouth disease in Tanzania', Bulletin of Epizootic Diseases of Africa 20, 101-104. http://dx.doi.org/10.1111/j.1865-1682.2007.01013.x [ Links ]
Rweyemamu, M.M. & Loretu, K., 1973, 'Observations on the first epidemic of foot-and-mouth disease Type SAT1 in Tanzania', Bulletin of Epizootic Diseases of Africa 21, 393-400. [ Links ]
Rweyemamu, M.M., Roeder P., MacKay D., Sumption K., Brownlie J., Leforban Y. et al., 2008, 'Epidemiological patterns of foot-and-mouth disease worldwide', Transboundary and Emerging Diseases 55(1), 57-72. [ Links ]
Samuel, A.R. & Knowles N.J., 2001, 'Foot-and-mouth disease type O viruses exhibit genetically and geographically distinct evolutionary lineages topotypes', Journal of General Virology 82, 609-621. [ Links ]
Tamura, K., Dudley, J., Nei, M. & Kumar, S., 2007. 'MEGA4: Molecular evolution genetics analysis (MEGA) software version 5.1', Molecular Biology Evolution 24, 1596-1599. http://dx.doi.org/10.1093/molbev/msm092 [ Links ]
Shaw, A.E., Reid, S.M., Ebert, K., Hutchings, G.H., Ferris, N.P. & King, D.P., 2007, 'Implementation of one step real-time RT-PCR protocol for diagnosis of foot-and-mouth disease', Journal of Virology Methods 143, 81-85. http://dx.doi.org/10.1016/j.jviromet.2007.02.009 [ Links ]
Swai, E.S., Mrosso, A. & Masambu, J.I.G., 2009, 'Occurrence of foot-and-mouth disease serotypes in Tanzania: A retrospective study of tongue epithelial tissue samples', Tanzania Veterinary Journal 26, 7-12. http://dx.doi.org/10.4314/tvj.v26i1.49232 [ Links ]
Vosloo, W., Bastos, A.D., Sangare, O., Hargreaves, S.K. & Thomson, G.R., 2002, 'Review of the status and control of foot-and-mouth disease in sub-Saharan Africa', Revue Scientifique et Technique (OIE) 21, 437-444. [ Links ]
 Correspondence:
Correspondence:
Raphael Sallu
PO Box 9254
Dar es Salaam, Tanzania
raphael.sallu@sacids.org
Note: Proceedings of the 2nd One Health Conference in Africa. Jointly organised by the Southern African Centre for Infectious Disease Surveillance and the Tanzania National Institute for Medical Research, held at the Snow Crest Hotel in Arusha, Tanzania from 16th to 19th April 2013: http://www.sacids.org/kms/frontend/index.php?m=119.


{kind=link}