










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkOnderstepoort Journal of Veterinary Research
versión On-line ISSN 2219-0635
versión impresa ISSN 0030-2465
Onderstepoort j. vet. res. vol.78 no.1 Pretoria ene. 2011
ORIGINAL RESEARCH
Molecular characterisation of Mycobacterium bovis isolated from African buffaloes (Syncerus caffer) in Hluhluwe-iMfolozi Park in KwaZulu-Natal, South Africa
Tiny M. HlokweI; Akinbowale O. JenkinsII; Elizabeth M. StreicherIII; Estelle H. VenterII; Dave CooperIV; Jacques GodfroidV; Anita L. MichelII
ITuberculosis Laboratory, ARC-Onderstepoort Veterinary Institute, South Africa
IIDepartment of Veterinary Tropical Diseases, University of Pretoria, South Africa
IIIDepartment of Biomedical Sciences, Stellenbosch University, South Africa
IVEzemvelo KZN Wildlife, St Lucia, South Africa
VDepartment of Food Safety and Infection Biology, Norwegian School of Veterinary Science, Norway
ABSTRACT
Bovine tuberculosis (BTB), a chronic disease of mammals caused by Mycobacterium bovis, is a threat to South African wildlife. It has been reported that African buffaloes (Syncerus caffer) are reservoir hosts of BTB in South African wildlife populations. This study reports on the molecular identification and typing of 31 M. bovis isolates collected between 1993 and 2008, mainly from buffaloes but also from two lions and a bush pig, in the Hluhluwe-iMfolozi Park (HiP) in KwaZulu-Natal. To study the dynamics of BTB in the buffalo populations, 28 M. bovis isolates from the HiP and epidemiologically related parks were characterised using regions of difference deletion analysis for species identification and spoligotyping, variable number of tandem repeats (VNTR), polymorphic G-C-rich sequences and IS6110 restriction fragment length polymorphism (RFLP) genotyping methods. At least three distinct M. bovis genotypes were found amongst HiP samples. The combination of VNTR typing (using a 16-loci panel) and IS6110 RFLP revealed the presence of three additional genetic profiles in individual buffaloes, demonstrating that the highest level of discrimination was achieved by these typing methods. One of the observed spoligotypes (SB0130) was dominant and represented 75% of isolates from buffaloes. A novel M. bovis spoligotype (SB1474), which is reported for the first time in this study, was observed in 14.3% of isolates from buffaloes. Based on the observed genetic relationships, the findings suggest independent introductions from at least three unrelated sources. These findings improve the knowledge regarding the diversity of circulating M. bovis strains in the HiP.
Introduction
Bovine tuberculosis (BTB) is a chronic, contagious disease caused by Mycobacterium bovis, a Gram-positive acid-fast bacterium. This pathogen is particularly noted for its diverse host tropism, which includes humans, livestock and several wildlife species. The first report of BTB in cattle in South Africa was made by Hutcheon (1880), whilst it was first diagnosed in African buffaloes (Syncerus caffer) in the Hluhluwe-iMfolozi Park (HiP) in 1986 (Jolles 2004). Spillover of BTB to other species has been documented (Michel et al. 2006; Michel et al. 2009). The HiP is situated in KwaZulu-Natal, South Africa and the area of 100 000 ha is almost entirely surrounded by communal farmland and livestock. It is the fourth largest game reserve in South Africa and has a buffalo population of approximately 3500 heads. The molecular characterisation of M. bovis is essential in understanding disease transmission between species and the spatial distribution of this infection in wildlife populations. In a previous study, molecular characterisation of M. bovis isolated from wildlife in the HiP was performed using a combination of three genetic tools, namely spoligotyping and restriction fragment length polymorphism (RFLP), including IS6110 and polymorphic G-C-rich sequence (PGRS) typing (Michel et al. 2009). In that study, M. bovis isolates, the majority of which were derived from buffaloes, had been collected in the HiP and two epidemiologically related parks between 1993 and 2000. With one exception (TB 328), which had a shift in one of the bands (Michel et al. 2009), the isolates all displayed the same IS6110 RFLP banding pattern. In addition, spoligotyping yielded only one type identical to SB0130 in the international M. bovis database (www.mbovis.org). This report also showed that epidemiologically unrelated strains are in circulation in the HiP and in the greater Kruger National Park (Michel et al. 2009).
Previous studies showed that variable number of tandem repeats (VNTR) typing is more discriminatory for M. bovis isolates than spoligotyping (Allix et al. 2006). In a study by Romero and co-workers (2008), both spoligotyping and VNTR were found to display similar discriminatory capacity. On the basis of the above information, the aim of this study was to investigate the circulating strains of M. bovis in the HiP further. Regions-of-difference (RD) deletion analysis was used for the identification of the isolates and spoligotyping, VNTR, PGRS and IS6110 RFLP techniques were used for genotyping.
Methods
Sample collection
Tissue samples, collected from tuberculin skin tests and/or buffaloes that had tested positive for gamma interferon during BTB surveys between 1993 and 2008, were transferred to the Tuberculosis Laboratory at the Onderstepoort Veterinary Institute (OVI) for bacterial culture. A total of 28 buffaloes, from the HiP (n = 20), Munyawana Game Reserve (n = 4), St Lucia Wetland game parks (n = 3) and a private game reserve (n = 1), were included in the study. Buffaloes from Munyawana and St Lucia Wetland game parks (both in KwaZulu-Natal) were originally from the HiP, but were moved to the new locations between 1997 and 2000. A buffalo from a private game reserve (situated in Mpumalanga Province) was born to a dam originating from the HiP. Additional samples included in this study were collected during ad-hoc post-mortem examinations from two lions (Panthera leo) from, respectively, the HiP and Munyawana Game Reserve, and a bush pig (Potamochoerus larvatus) from the HiP.
Bacterial isolation and molecular identification
The tissue samples were processed at the Tuberculosis Laboratory, OVI, according to standard procedures (Alexander et al. 2002; Bengis et al. 1996). The samples were inoculated onto Löwenstein-Jensen media supplemented with pyruvate and incubated at 37 ºC for up to 10 weeks. Acid-fast bacterial isolates were subjected to polymerase chain reaction (PCR) amplification using primers that target a sequence encoding the MPB 70 antigen to identify Mycobacterium tuberculosis complex bacteria (Alexander et al. 2002; Bengis et al. 1996; Michel et al. 2009). M. tuberculosis complex bacteria were identified following the presence of a 372-bp PCR product. Deletion analysis was performed as described by Warren et al. (2006) for species identification. The RD4 and RD9 genomic regions of difference were targeted using a multiplex PCR approach (Warren et al. 2006). The PCR products were separated on a 2-3% agarose gel to allow for good definition of the bands. M. bovis isolates were confirmed by the presence of two specific bands of 268 bp and 108 bp for RD4 and RD9, respectively.
Genomic DNA extraction
A PUREGENETMDNA extraction kit was used to extract genomic DNA from isolates according to the manufacturer's instructions (Gentra Systems, Minneapolis), with minor modifications. Cells treated overnight with glycine were heat killed at 94 ºC for 10 minutes and allowed to cool. A volume of 500 µL extraction buffer (50 g/L monosodium glutamic acid; 6.06 g/L Tris-HCl (pH 7.4); 9.3 g/L EDTA) was added to the culture and cells were suspended by using an inoculating loop. Following centrifugation, 300 µL lysis solution and 5 µL RNase A solution (4 mg/mL) (Roche Diagnostics, Randburg) were added and the sample was then incubated at 37 ºC for 30 min. A volume of 150 µL protein precipitation solution was added and samples were centrifuged at maximum speed for 2 min, followed by precipitation of the aqueous phase with isopropanol (Merck, Halfway House). The resulting pellet was dissolved in 40 µL TE buffer (10 mM Tris-HCl; 1 mM EDTA). The extracted DNA was stored at -20 ºC until use.
Genotyping
Either pure genomic DNA (extracted using the PUREGENETM DNA extraction kit) or cell lysates (prepared by boiling the M. bovis cells for 25 min and storing at -20 ºC until used) were used for the PCR-based typing methods (i.e. spoligotyping and VNTR typing). For RFLP typing, pure genomic DNA was used.
Spoligotyping
Spoligotyping was done according to a standardised international method (Kamerbeek et al. 1997) using a commercially available kit (Isogen Biosciences BV, Maarsen). M. tuberculosis H37Rv, M. bovis BCG and sterile distilled water were used as controls. The spoligotype patterns were compared to those stored in the M. bovis spoligotype database (www.mbovis.org).
Variable number of tandem repeats typing
Cell lysates or genomic DNA was used for PCR amplification. Initially, VNTR analysis, also called multiple locus variable analysis, was carried out using primers to amplify the ETR-A to ETR-F loci (Frothingham & Meeker-O'Connell 1998). Additional VNTR typing was performed on 10 loci (Mtub 2, Mtub 12, Mtub 21, Mtub 29, Mtub 30, Mtub 39, MIRU 2, MIRU 10, MIRU 16 and Qub 11a) as described by Le Fleche et al. (2002). PCR was performed using an Eppendorf AG 22331 Hamburg thermocycler (Merck Eppendorf, Hamburg). The PCR products were separated on a 2% agarose gel at 85 V for 3 h and visualised under 312 nm UV light (Spectroline® model TC-312A transilluminator, Westbury). A 100-bp ladder (Inqaba Biotechnology, Pretoria) was included as a marker. The band sizes were determined using Quantity One® 1-D analysis software installed in the Gel Doc system (Bio-Rad Laboratories, Hercules, CA), converted to copy numbers (for exact tandem repeats [ETRs], see Frothingham & Meeker-O'Connell 1998; for additional loci, see Le Fleche et al. 2002) and saved in a spreadsheet. The genotyping data were exported to Bionumerics software (Applied Maths, Sint-Martens-Latem). The categorical coefficient option was used for cluster analysis to create a dendogram.
Polymorphic G-C-rich sequence and IS6110 typing
PGRS and IS6110 RFLP typing were performed as previously reported (Cousins et al. 1998; Michel et al. 2008). The GelCompar II system (Applied Maths, Sint-Martens-Latem) was used for analysis of the IS6110 RFLP results, with the similarity coefficient of Dice and the unweighted pair group method with arithmetic averages for clustering maximum tolerance at 1.2%. PGRS RFLP typing patterns results were analysed manually.
Results
Mycobacterium bovis isolation and molecular identification
Bacterial growth was observed on Löwenstein-Jensen culture slopes after 6-10 weeks of incubation. The presence of acid-fast bacilli was confirmed by means of Ziehl-Neelsen stains. Isolates were classified as members of the M. tuberculosis complex following detection of a 372-bp fragment by PCR using primers targeting the sequence that codes for the MPB 70 antigen of the M. tuberculosis complex. Deletion analysis using multiplex PCR targeting RD4 and RD9 (Warren et al. 2006) further confirmed the isolates as M. bovis owing to the amplification of DNA products with band sizes of 268 bp and 108 bp, respectively (results not shown).
Spoligotyping
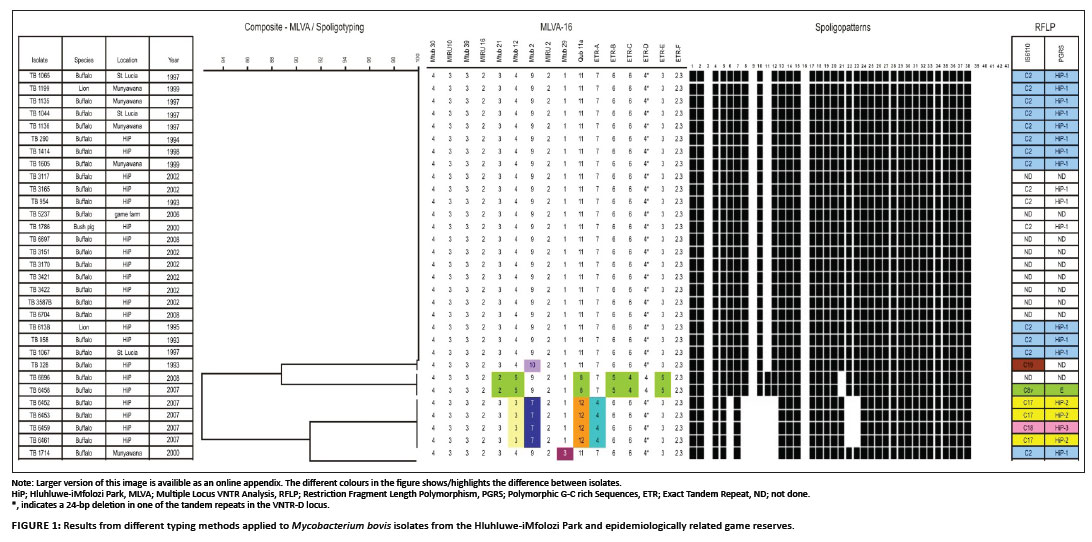
Spoligotyping was performed on all 31 M. bovis isolates: 28 from buffaloes; two from lions and one from a bush pig. The results (Figure 1 and Figure 2) revealed four different spoligopatterns. The most prevalent spoligotype pattern was found in 24 of the 31 (77.4%) M. bovis isolates, including 21 isolates from buffaloes, two from lions and that of the bush pig. This spoligotype pattern was marked by the absence of spacers 3, 9, 11, 16 and 39-43, which is consistent with SB0130 (www.mbovis.org). The strain type was identified in isolates collected from the HiP during the study period (1993-2008) and has been reported earlier (Michel et al. 2009). A second spoligotype pattern was found in four buffaloes (TB 6452, TB 6453, TB 6459 and TB 6461) sampled in 2007. This M. bovis spoligopattern has not been described in the M. bovis database before and was allocated a new code, SB1474. It is characterised by the lack of spacers 3, 6, 8, 9, 10, 11, 12, 16, 22, 23 and 39-43. A third spoligotype pattern was found in two M. bovis isolates from buffaloes sampled in 2007 and 2008 (TB 6458 and TB 6696, respectively). The pattern, designated SB0121, is delineated by the absence of spacers 3, 9, 16, 21 and 39-43. This pattern is similar to that for M. bovis BCG except that spacer 21 is present in the latter. Another spoligotype pattern was isolated in 2000 from a buffalo (TB 1714) that was translocated from the HiP and was kept in a boma at Munyawana Game Reserve. This particular pattern was characterised by the absence of spacers 3, 6, 8, 9, 10, 11, 12, 16 and 39-43. It is identical to SB0140 in the international M. bovis database.
Analysis of variable number of tandem repeats
Results of the VNTR analyses on all 31 isolates, using a total of 16 loci, are summarised in Figure 1. Comparison of the copy numbers of the VNTR profile of each isolate revealed five profiles. The most prevalent VNTR profile was found in 23 of the 31 M. bovis isolates from buffaloes, lions and a bush pig. This VNTR profile was essentially shared by TB 328 (second profile) and TB 1714 (third profile), except for the copy numbers relating to locus Mtub 12 (in TB 328) and Mtub 29 (in TB 1714). The remaining two VNTR profiles were found in four (TB 6452, TB 6453, TB 6459, TB 6461) and two M. bovis isolates from buffaloes (TB 6458 and TB 6696) collected in 2007 and 2008, respectively). The genetic diversity observed in these isolates were attributed to the use of a combination of the loci ETR-A, ETR-B, ETR-C, ETR-E, Mtub 2, Mtub 12, Mtub 21, Mtub 29 and Qub 11a.
Polymorphic G-C-rich sequences
PGRS typing was performed on five M. bovis isolates cultured from buffaloes sampled in 2007 (Figure 1: TB 6452, TB 6453, TB 6459, TB 6461 and TB 6458). The PGRS profiles of 16 isolates have been analysed previously and were found to share a unique profile (Michel et al. 2009). In the present study, three PGRS DNA profiles were observed. One profile (HiP-2) was present in three of the five M. bovis isolates analysed, whilst the two remaining isolates each exhibited a genetically different profile (HiP-3 and E, respectively) as shown in Figure 1.
IS6110 typing
The same five M. bovis isolates as analysed by PGRS typing were subjected to IS6110 typing. Three IS6110 patterns were identified, namely C17, C18 and C8v. M. bovis isolates with identical PGRS patterns also displayed identical IS6110 patterns (Figure 1).
Discussion
This study presents data obtained from M. bovis isolates from the HiP and three epidemiologically related game reserves. PCR-based genotyping tools (i.e. spoligotyping and VNTR typing) were primarily applied to characterise 31 M. bovis isolates, complemented with PGRS and IS6110 typing on some of the samples for added discriminatory power.
Most M. bovis infections examined in this study were caused by a unique genotype, which corresponded to a spoligopattern that had previously been identified as SB0130 (Michel et al. 2009). These results were in line with those from VNTR typing, except for VNTR typing singling out one isolate (TB 328) on the basis of a difference in locus Mtub 2. This finding was supported by previous IS6110 typing results (Michel et al. 2009). Based on the high degree of homology between TB 328 and the dominant VNTR typing profile, it is unlikely that TB 328 resulted from a separate introduction of M. bovis into the HiP; rather, it may be linked to the dominant strain through an evolutionary process.
Spoligotyping and VNTR typing results were also found to correspond to a unique genotype isolated in buffaloes sampled in 2007 (TB 6452, TB 6253, TB 6459, TB 6461). The spoligopattern has not been described in the HiP previously nor included in the international M.bovis database and was therefore designated a new code, SB1474. However, when IS6110 and PGRS typing data were considered for this group of isolates, both markers were able to discriminate between two profiles (e.g. C17 and C18). The genetic diversity of these strains collected in 2007 suggests that they did not evolve from the dominant HiP strain (SB0130) and therefore most likely represent an independent introduction of M. bovis into the HiP.
During the period prior to 2000 buffalo TB 1714 was translocated from the HiP to Munyawana Game Reserve where it was euthanised and sampled in 2000. Spoligotyping revealed a strain, SB0140, which is related to the novel SB1474. The novel strain (SB1474) differs from SB0140 by deletion of spacers 22 and 23 in addition to the loss of spacers 3, 6, 8, 9, 10, 11, 12 and 16, which is descriptive of the BOVIS2 family (Brudey et al. 2006). The origin of strain SB0140 remains elusive, despite identical PGRS and IS6110 typing results with SB0130 as shown in Figure 1.
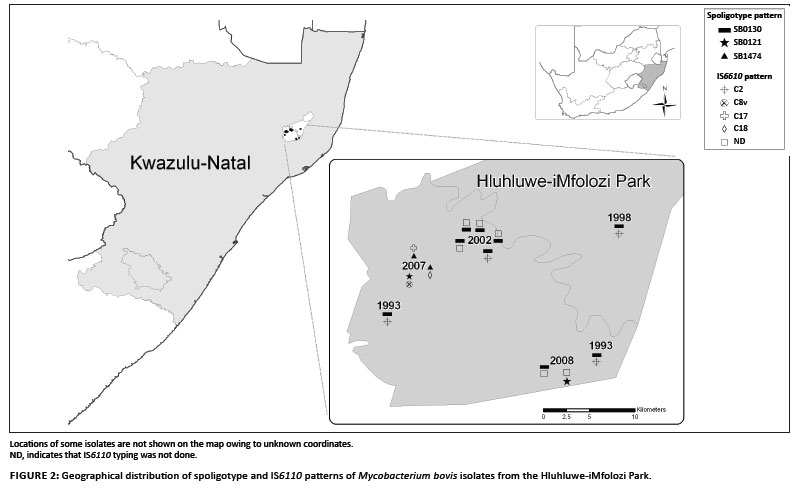
Evidence of another independent source of M. bovis in the HiP was found in two buffaloes (TB 6458 and TB 6696) sampled in 2007 and 2008, respectively, which both carried the spoligotype SB0121. This particular spoligotype pattern was previously isolated in cattle in Gauteng, Mpumalanga and the Limpopo provinces of South Africa (Michel et al. 2008), as well as in buffaloes and other wildlife in the Kruger National Park (Michel et al. 2009). Additional IS6110 and PGRS typing (only on TB 6458) confirmed that this genotype was a member of the C8 variant strain complex, which is the cause of the BTB epidemic in the Kruger National Park (Michel et al. 2009). It is unclear whether the strain could have been introduced more recently from infected cattle or wildlife translocated from areas neighbouring the Kruger National Park to areas neighbouring the HiP. Alternatively, the C8 strain as found in the Kruger National Park may have been more widely spread in the eastern parts of the country than previously known and undergone similar evolutionary processes as described for the Kruger National Park (Michel et al. 2009). It is also possible that, at a particular time, strains present in buffaloes sampled in 2007 (western part of the HiP) and 2008 (eastern part of the HiP) may not have been present in buffaloes sampled before 2002 (eastern part of the HiP). Indeed, earlier behavioural studies indicate that HiP buffaloes have relatively stable and small home ranges (compared to those in the Kruger National Park) owing to the abundant year-round food and water availability in the HiP (Dora 2004). Very little mixing, if any, of breeding herds is known, with adult bulls implicated as the main source of infection among herds.
The use of molecular typing methods in the present study proved to be useful in confirming the origins of M. bovis strains translocated to, for example, St Lucia Wetland game parks in KwaZulu-Natal and a private game reserve in Mpumalanga. In all cases the M. bovis strain types were identical to the most prevalent type in the HiP. The methods show that the prevalent strain has spilled over to wildlife species other than buffaloes, as evidenced by the cases of two lions and a bush pig in the present study, and by IS6110 typing in a previous study by Michel and co-workers (2009).
The identification of previously undetected M. bovis strains in the HiP is of great concern, since it indicates the possibility of multiple introductions in the past and a persistent risk for new M. bovis introductions into the park, as well as a risk of spillover to surrounding communal cattle farms. This presents a challenge for controlling the disease in livestock and has an impact on wildlife management and potentially also on public health (Michel et al. 2006; Romero et al. 2008).
Conclusion
The molecular typing methods described here have been useful in studying the epidemiology of BTB in the HiP and provide valuable information for future disease management strategies. This study further suggests that detection surveys for BTB are needed in communal cattle herds around the HiP to identify and efficiently manage sources of M. bovis infection.
Acknowledgements
We thank Dr Noel Smith and staff at the Veterinary Laboratory Agency for VNTR analysis training and providing the VNTR A, B, C, E and F primers, Gilles Vergnaud of the Institute of Genetics and Microbiology, University of Paris XI Orsay, also for VNTR analysis training, Dr Roy Williams for creating the map used in Figure 2, and Dr Claude Sabeta for critically reviewing the manuscript. The work was funded jointly by the Department of Agriculture, the Department of Science and Technology, the Belgian Directorate-General for Development Cooperation (Research project 9740X) and the University of Pretoria.
Author's contributions
T.M.H. and A.O.J. contributed equally to the design of the study and preparation of the manuscript. E.M.S. performed spoligotyping of isolates. D.C. provided information relating to animals involved and their locations. A.L.M., J.G. and E.H.V. were involved in drafting the manuscript and revising it critically. All authors read and approved the final manuscript.
References
Alexander, K.A., Pleydell, E., Williams, M.C., Lane, E.P., Nyange, J.F.C. & Michel, A.L., 2002, 'Mycobacterium tuberculosis: an emerging disease of free-ranging wildlife', Emerging Infectious Disease 8, 592-595. [ Links ]
Allix, C., Walravens, K., Saegerman, C., Godfroid, J., Supply, P. & Fauville-Dufaux, M., 2006, 'Evaluation of the epidemiological relevance of variable-number tandem-repeat genotyping of Mycobacterium bovis and comparison of the method with IS6110 restriction fragment length polymorphism analysis and spoligotyping', Journal of Clinical Microbiology 44, 1951-1962. doi:10.1128/JCM.01775-05, PMid:16757584, PMCid:1489394 [ Links ]
Bengis, R.G., Kriek, N.P.J., Keet, D.F., Raath, J.P., De Vos, V. & Huchzermeyer, H.F.K.A., 1996, 'An outbreak of bovine tuberculosis in a free-living buffalo population in the Kruger National Park', Ondestepoort Journal of Veterinary Research 63, 15-18. PMid:8848298 [ Links ]
Brudey, K., Driscoll, J.R., Rigouts, L., Prodinger, W.M., Gori, A., Al-Hajoj, S.A. et al., 2006, 'Mycobacterium tuberculosis complex genetic diversity: Mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology', BMC Microbiology 6, 23. doi:10.1186/1471-2180-6-23, PMid:16519816, PMCid:1468417 [ Links ]
Cousins, D., Williams, S., Liebana, E., Aranaz, A., Bunschoten, A., Van Embden, J. et al., 1998, 'Evaluation of four DNA typing techniques in the epidemiological investigations of bovine tuberculosis', Journal of Clinical Microbiology 36, 168-178. PMid:9431942, PMCid:124829 [ Links ]
Dora, C.A., 2004, 'The influences of habitat structure and landscape heterogeneity on African buffalo (Syncerus Caffer) group size in Hluhluwe-iMfolozi Park, South Africa', MSc thesis, University of Oregon. [ Links ]
Frothingham, R. & Meeker-O'Connell, W.A., 1998, 'Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats', Microbiology 144, 1189-1196. doi:10.1099/00221287-144-5-1189, PMid:9611793 [ Links ]
Hutcheon, D., 1880, 'Tering, consumption, tables mesenterica', Annual Report, Colonial Veterinary Surgeon, Cape of Good Hope. [ Links ]
Jolles, A., 2004, 'Disease ecology of bovine tuberculosis in African buffalo', PhD thesis, Department of Ecology and Evolutionary Biology, Princeton University. [ Links ]
Kamerbeek, J., Schouls, L., Kolk, A., Van Agterveld, M., Van Soolingen, D., Kuijper, S. et al., 1997, 'Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology', Journal of Clinical Microbiology 35, 907-914. PMid:9157152, PMCid:229700 [ Links ]
Le Fleche, P., Fabre, M., Denoeud, F., Koeck, J.L. & Vergnaud, G., 2002, 'High resolution, on-line identification of strains from the Mycobacterium tuberculosis complex based on tandem repeat typing', BMC Microbiology 2, 37. doi:10.1186/1471-2180-2-37, PMCid:140014 [ Links ]
Michel, A.L., Bengis, R.G., Keet, D.F., Hofmeyr, M., De Klerk, L.M., Cross, P.C. et al., 2006, 'Wildlife tuberculosis in South African conservation areas: implications and challenges', Veterinary Microbiology 112, 91-100. doi:10.1016/j.vetmic.2005.11.035, PMid:16343819 [ Links ]
Michel, A.L., Hlokwe, T.M., Coetzee, M.L., Mare, L., Connoway, L., Rutten, V.P. et al., 2008, 'High Mycobacterium bovis genetic diversity in a low prevalence setting', Veterinary Microbiology 126, 151-159. doi:10.1016/j.vetmic.2007.07.015, PMid:17720336 [ Links ]
Michel, A.L., Coetzee, M.L., Keet, D.F., Maré, L., Warren, R., Cooper, D. et al., 2009, 'Molecular epidemiology of Mycobacterium bovis isolates from free-ranging wildlife in South African game reserves', Veterinary Microbiology 133, 335-343. doi:10.1016/j.vetmic.2008.07.023, PMid:18786785 [ Links ]
Romero, B., Aranaz, A., Sandoval, A., Alvarez, J., de Juan, L., Bezos, J. et al., 2008, 'Persistence and molecular evolution of Mycobacterium bovis population from cattle and wildlife in Donana National Park revealed by genotyping variation', Veterinary Microbiology 132, 87-95. doi:10.1016/j.vetmic.2008.04.032, PMid:18539410 [ Links ]
Warren, R.M., Gey van Pittius, N.C., Barnard, M., Hesseling, A., Engelke, E., De Kock, M. et al., 2006, 'Differentiation of Mycobacterium tuberculosis complex by PCR amplification of genomic regions of difference', International Journal of Tuberculosis Lung Disease 10, 818-822. PMid:16850559 [ Links ]
 Correspondence to:
Correspondence to:
Tiny Hlokwe
Postal address: Private Bag X05
Onderstepoort 0110, South Africa
Email: HlokweT@arc.agric.za
Received: 20 Sept. 2010
Accepted: 19 Jan. 2011
Published: 20 June 2011
© 2011. The Authors. Licensee: OpenJournals Publishing. This work is licensed under the Creative Commons Attribution License.


{kind=link}
{kind=link}