










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Dental Journal
versión On-line ISSN 0375-1562
versión impresa ISSN 0011-8516
S. Afr. dent. j. vol.75 no.9 Johannesburg oct. 2020
http://dx.doi.org/10.17159/2519-0105/2020/v75no9a3
CASE REPORT
Granulomatosis with polyangiitis - An oral medicine case book
I SnymanI; L RobinsonII; AW van ZylIII; WFP van HeerdenIV
IBChD, PDD (Implantology), PGDipDent (Oral Surgery), PGDipDent (Implantology), MChD, FCD (SA) OMP, Private Practice Periodontist, Stellenbosch, South Africa. ORCID Number: 0000-0002-8480-5361
IIBChD, PDD (Maxillofacial Radiology), PDD (Forensic Odontology), Department of Oral Pathology and Oral Biology, School of Dentistry, Faculty of Health Sciences, University of Pretoria, South Africa. ORCID Number: 0000-0002-0549-7824
IIIBChD, MChD, Private Practice Periodontist, Hermanus & Honorary Professor, Department of Oral Medicine and Periodontics, Faculty of Health Sciences, University of Wit-watersrand, South Africa. ORCID Number: 0000-0002-7985-4054
IVBChD, MChD, FC Path (SA) Oral Path, PhD, DSc, Department of Oral Pathology and Oral Biology, School of Dentistry, Faculty of Health Sciences, University of Pretoria, South Africa. ORCID Number: 0000-0003-2494-667X
CASE REPORT
A 33-year-old female patient presented at a private specialist practice (Periodontics and Oral Medicine) with a main complaint of sore and bleeding gums, fever, headaches and sinus pain. The gingiva became irritated, red and swollen five weeks prior to the first consultation in July 2018.
She was referred by her dentist one month after prescribing two consecutive courses of antibiotics and providing oral hygiene instructions to the patient, with no significant improvement. The first prescription was for Augmentin 375mg t.d.s. for 5 days and the second prescription two weeks later was for Amoxycillin 250 mg t.d.s. and Metronidazole 200mg t.d.s. for 5 days. The patient was also using a chlorhexidine mouth rinse.
The patient's medical history included the following:
• Caesarian section 8-months prior to consultation.
• Quit smoking (5-10 cigarettes/day) before conceiving in 2017.
• Plantar psoriasis of the left foot, diagnosed after clinical examination by a Rheumatologist.
• Childhood allergic rhinitis (hay fever).
• No known allergies.
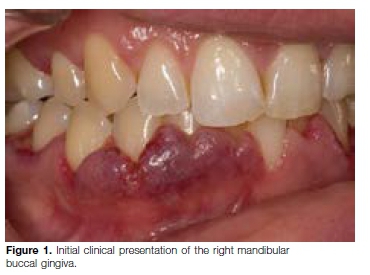
On examination, the gingiva appeared erythematous and swollen with a "strawberry appearance". Both the buccal and lingual gingiva in the upper and lower jaws were affected (Figures 1 & 2). The affected areas of the gingi-va were firm, tender and exhibited spontaneous bleeding at times. At this stage, the differential diagnosis included granulomatosis with polyangiitis, pyostomatitis vegetans or a leukemic infiltrate.

DIAGNOSIS AND MANAGEMENT
An incisional biopsy was performed with a scalpel (15C blade) under local anaesthesia of the affected gingiva, lingual to the left mandibular first molar, for histologi-cal examination. Sutures were placed (chromic gut 4/0), post-operative instructions were given and healing was uneventful.
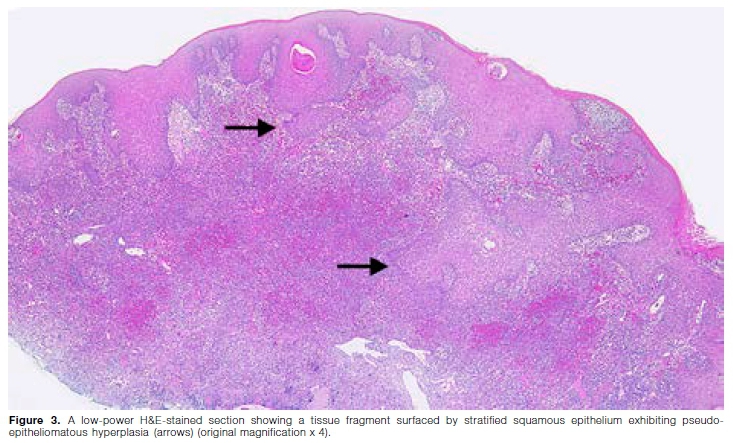
Macroscopic examination revealed a specimen consisting of a scalloped fragment of mucosa with a granular surface, measuring 29x8x3mm. Histological evaluation showed a tissue fragment surfaced by acanthotic, hyperparakeratotic stratified squamous epithelium exhibiting pseudoepitheliomatous hyperplasia and moderate-to-severe inflammatory exocytosis (Figure 3). The underlying lamina propria contained large areas of suppuration with microabscess formation. Numerous poorly formed granulomas composed of lymphocytes and histiocytes were noted, with eosinophils and plasma cells present throughout the adjacent stroma. Prominent multinucleated giant cells, varying in size and shape and containing 5 to 15 nuclei, were present throughout the lesion (Figure 4). There were abundant capillaries involved by a leukocytoclastic vasculitis with associated erythrocyte extravasation and fibrin deposition. There was no evidence of epithelial dysplasia or malignancy in the sections examined. Periodic acid-Schiff (PAS), Warthin-Starry and Ziehl-Neelsen histochemical stains failed to highlight any fungal elements, bacilli/ spirochetes or acid-fast bacilli respectively. These his-tological features were in keeping with granulomatosis with polyangiitis.
The patient was referred to a Rheumatologist for a comprehensive systemic evaluation. Multiple blood tests were ordered and the results recorded in Table 1. The patient met the American College of Rheumatology criteria according to the 1990 classification for granu-lomatosis with polyangiitis:1

1. Oral inflammation.
2. Abnormal urinary sediment.
3. Granulomatous inflammation upon biopsy.
The clinical, histological and laboratory findings confirmed a final diagnosis of granulomatosis with polyangiitis.
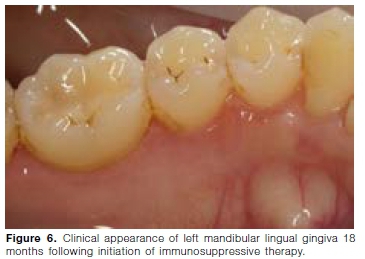
Initial treatment, starting in July 2018, consisted of systemic prednisone (5mg/day) and azathioprine (50mg/ day). After six months, the response to treatment was excellent, however it was not possible to wean the patient off prednisone. Azathioprine was ceased and Methotrexate (20mg/week) initiated in January 2019. A renal biopsy in June 2019 showed mild mesangial proliferative glomerulonephritis. During a follow-up visit in December 2019, no gingival lesions were noted (Figures 5 and 6). The urine protein/creatinine ratio was normal and the psoriatic skin lesion on the left foot improved. Her respiratory examination was clear and she had no active joint disease. She was still very sensitive to prednisone withdrawal and her treatment was adjusted as follows:

• Systemic prednisone 2,5 mg on alternate days.
• Methotrexate 20 mg weekly.
• Folate 5 mg weekly.
• Caltrate Plus 1 tablet daily.
• Cardiac Aspirin 1 tablet on alternate days.
DISCUSSION
Since the shift from honorific eponyms to more disease descriptive or aetiology-based nomenclature, Wegener's granulomatosis has been renamed as granulomatosis with polyangiitis (GPA).2 The disease was first described by Klinger in 1931, and only later by Friedrich Wegener as a separate syndrome.2,3,4,5 An annual incidence of 5-10 cases per million population has been reported.6,7 GPA is usually diagnosed in individuals between 45 and 60 years of age, and is very rare in children.6,8 Males and females are equally affected with the disease being more prevalent amongst caucasian individuals than other population groups.6,8,7,9
According to the 2012 International Chapel Hill Consensus Conference on the Nomenclature of Vasculitides, GPA is defined as a systemic necrotising granulomatous inflammatory disease which usually involves the upper and lower respiratory tract, with necrotising vasculitis affecting predominantly small and medium-sized blood vessels.9,10 The acronym 'ELK' is used to describe clinical involvement of the ear, nose and throat; lungs and kidneys.11 GPA may only present locoregionally, as a limited form of the disease, or progress to a more widespread systemic form if left untreated.11 GPA may affect the skin, oral cavity, eyes, nose and paranasal sinuses, ears, upper and lower airway, cardiovascular system, gastrointestinal system, kidneys, central and peripheral nervous system and musculoskeletal system.7,11
The precise cause(s) of GPA remain(s) unknown.12 The disease likely represents a form of T-cell-mediated hypersensitivity response to normal "innocuous" inhaled microbial or other environmental agents.12 Environmental factors, such as silica or dust inhalation have been implicated in a minority of patients as a possible trigger.8 Infectious agents may also play a role through the process of molecular mimicry.8 The role of genetic factors was also demonstrated in a genome-wide association study.13 The HLA class II region is the best-established risk gene/locus in GPA, with HLA-DPB1 and HLA-DPA1 variants showing the strongest association signals.14 B-cells also possibly play a major role in the patho-genesis as they produce anti-neutrophil cytoplasmic antibodies (ANCAs). Anti-proteinase-3 antibodies (PR3-ANCA, previously c-ANCA) are present in up to 95% of cases, and may participate in the disease pathogenesis.12 GPA, along with microscopic polyangiitis and eosinophilic granulomatosis with polyangiitis (Churg-Strauss) are collectively referred to as ANCA-associated vasculitides.14 The presence of these antibodies and their titers appear to be related to levels of disease activity. Imbalances in different T-cell subtypes and/or cytokine-chemokine networks can also lead to or at least participate in the failure of tolerance, triggering auto-immunity.12
Orofacial manifestations of GPA include labial mucosal nodules, gingival enlargement, granulomatous lesions, oral ulceration, extraction sockets that fail to heal, oro-antral fistulae, temporomandibular joint arthralgia, facial nerve palsy and salivary gland enlargement.15,16 A chronic destructive process of the nasal cavity may lead to palatal ulcers.15 Nasal septum collapse may lead to a saddle nose deformity. The most characteristic oral manifestation of GPA is enlarged, red to purple gingiva, with a granular appearance referred to as strawberry gingivitis, because it resembles an over-ripe strawberry.15 Gingival GPA usually first involves the labial or lingual interdental areas and may progress to involve the entire buccal and/or lingual gingivae.16 Osteomyelitis and necrosis of the underlying bone may arise with subsequent mobility and tooth loss.16
The diagnosis of GPA is based on clinical assessment, serological testing and histological findings.11 The American College of Rheumatology proposed four diagnostic criteria, with a minimum of two required for a diagnosis of GPA (Table 2).1 The presence of any 2 or more criteria yields a sensitivity of 88.2% and a specificity of 92.0%.1 Histologically, GPA appears as a pattern of mixed inflammation, vasculitis, necrosis and poorly formed granulo-mas. All stages of vasculitis may be present, ranging from acute to granulomatous to healed. Extravasated erythrocytes and fibrin thrombi are often seen in association with damaged blood vessels. The adjacent connective tissue contains a variable mixture of histiocytes, lymphocytes, eosinophils and multinucleated giant cells. Areas of geographic coagulative necrosis may be present, with a prominent rim of palisaded epithelioid histiocytes. Giant cells may also be present around the necrotic areas. Special histochemical stains are negative for organisms, and no foreign material can be detected.8,13,17

Since the introduction of immunosuppressive therapy, the prognosis of GPA has greatly improved from a mean survival time of 5 months to a 10-year survival rate of approximately 80%.8,16 A diagnosis of GPA always necessitates treatment and is mainly guided by disease activity and severity.16 Therapy may include the use of corticos-teroids, cyclophosphamide, methotrexate, azathioprine, leflunomide, co-trimoxazole, mycophenolate, cyclosporine and rituximab.16 A combination of azathioprine and low-dose prednisone is mainly used as maintenance ther-apy.18 Relapse might occur in 50% of patients following remission, due to discontinuation of therapy, rapid tapering of corticosteroids or persistent positive-ANCA during the remission period.16 It should be emphasised that the management of GPA requires a multidisciplinary approach that may involve the dentist, oral medicine specialist, oral pathologist, maxillofacial surgeon, rheumatologist, pulmo-nologist, nephrologist and/or internal medicine specialist.7
References
1. Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990; 33(8): 1101-7. [ Links ]
2. Falk RJ, Gross WL, Guillevin L, et al. Granulomatosis with Polyangiitis (Wegener's): An alternative name for Wegener's Gran-ulomatosis. Arthritis Rheum. 2011; 63(4): 863-4. [ Links ]
3. Klinger H. Grenzformen der periarteritis nodosa. Frankfurt Z Path. 1931; 29: 455-80. [ Links ]
4. Wegener F. Über generalisierte, septische Gefásserkran-kungen. Verh dtsch pathol ges. 1936; 29: 202-9. [ Links ]
5. Wegener F. Über eine eigenartige rhinogene Granulomatose mit besonderer Beteilgung des Arterien systems under der Nieren. Beitr Pathol Anat. 1939; 102: 36-68. [ Links ]
6. Lane SE, Watts R, Scott DG. Epidemiology of systemic vascu-litis. Curr Rheumatol Rep. 2005; 7(4): 270-5. [ Links ]
7. Szczeklik K, Wtudarczyk A, Wawrzycka-Adamczyk K, et al. Oral manifestations of granulomatosis with polyangiitis -Clinical and radiological assessment. J Dent Sci. 2019; 14(1): 54-60. [ Links ]
8. Comarmond C, Cacoub P. Granulomatosis with polyangi-itis (Wegener): clinical aspects and treatment. Autoimmun Rev. 2014; 13(11): 1121-5. [ Links ]
9. Kertesz T, Soowamber M, Bubola J, Psutka DJ, Bradley G. Gingival Swelling as the Initial Manifestation of Granuloma-tosis with Polyangiitis. Head Neck Pathol. 2020; 1-10. [ Links ]
10. Jennette JC, Falk R, Bacon P, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vas-culitides. Arthritis Rheum. 2013; 65(1): 1-11. [ Links ]
11. Greco A, Marinelli C, Fusconi M, et al. Clinic manifestations in granulomatosis with polyangiitis. SAGE Publications Sage UK: London, England. 2016. [ Links ]
12. Pagnoux C. Updates in ANCA-associated vasculitis. Eur J Rheumatol. 2016; 3(3): 122. [ Links ]
13. Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012; 367(3): 214-23. [ Links ]
14. Xie G, Roshandel D, Sherva R, et al. Association of granulomatosis with polyangiitis (Wegener's) with HLA-DPB1*04 and SEMA6A gene variants: evidence from genome-wide analysis. Arthritis Rheum. 2013; 65(9): 2457-68. [ Links ]
15. Msallem B, Bassetti S, Matter MS, Thieringer FM. Strawberry gingivitis: Challenges in the diagnosis of granulomatosis with polyangiitis on gingival specimens. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019; 128(6):e202-e7. [ Links ]
16. Almouhawis HA, Leao JC, Fedele S, Porter SR. Wegener's granulomatosis: A review of clinical features and an update in diagnosis and treatment. J Oral Pathol Med. 2013; 42(7): 507-16. [ Links ]
17. Muller K, Lin JH. Orbital granulomatosis with polyangiitis (Wegener granulomatosis): clinical and pathologic findings. Arch Pathol Lab Med. 2014; 138(8): 1110-4. [ Links ]
18. Ponniah I, Shaheen A, Shankar K, Kumaran M. Wegener's granulomatosis: The current understanding. Oral Surg Oral Med Oral Pathol Oral Radiol Oral Endo. 2005; 100(3): 265-70. [ Links ]
 Correspondence:
Correspondence:
Willie FP van Heerden
Department of Oral Pathology and Oral Biology, University of Pretoria, South Africa.
Email: willie.vanheerden@up.ac.za
Author contributions:
1. Inus Snyman: Primary author - 40%
2. Liam Robinson: Secondary author - 30%
3. André W van Zyl: Clinical management and review - 15%
4. Willie FP van Heerden: Histological diagnosis and review - 15%


{kind=link}
{kind=link}