










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Dental Journal
versão On-line ISSN 0375-1562
versão impressa ISSN 0011-8516
S. Afr. dent. j. vol.70 no.2 Johannesburg Mar. 2015
CASE BOOK
Oral medicine case book 67: Oral manifestations of Evans syndrome: a presenting feature of HIV infection?
S RanchodI; A JefthaII; M MeyerIII; WP DreyerIV
IBChD, PDD. Department of Maxillofacial and Oral Surgery, Faculty of Dentistry, University of the Western Cape
IIBChD, MChD. Department of Oral Medicine and Periodontics, Faculty of Dentistry, University of the Western Cape
IIIBChD, HonsBScDentSci, DipOdont. Department of Maxillofacial and Oral Surgery, Faculty of Dentistry, University of the Western Cape
IVBDS, HDipDent, PhD, FCD(SA)OMP. Division of Oral Medicine and Periodontics, Faculty of Dentistry, University of the Western Cape; Professor Emeritus, Stellenbosch University
CASE REPORT
A 19 year old female presented with spontaneous intra-oral bleeding of two days duration. The patient reported that she was, until recently, in good general health and also that she had an uncomplicated parturition three years ago. She recently started noticing blood in her stools and felt increasingly lethargic. There was no history of trauma or intra-oral intervention that may have initiated the bleeding.
The clinical examination revealed marked pallor of the facial skin and multiple small petechiae were seen on both of her forearms. The intra-oral examination identified marked halitosis and multiple haemorrhagic lesions with a variable appearance, being plaque-like on the lip, nodular on the tongue and fungating and exophytic on the palate and in the retromolar regions. Even delicate manipulation of the tissues produced profuse bleeding (Figures 1-3).



Due to her general weakness and the excessive bleeding she was admitted to the local tertiary hospital with the suspicion of thrombocytopenia and possible HIV infection. The reason for the latter suspicion was that some of the oral lesions clinically resembled Kaposi sarcoma. Upon admission, the following vital signs were recorded: blood pressure (BP) 97/52; heart rate 140 beats/min; temperature 36°C; random blood glucose 8.7mmol/L; ward haemoglobin (Hb) 6.4g/dL; and weight 55kg. Venous blood was drawn and submitted for a full blood count, INR, direct Coombs test and test for HIV infection. There was a marked difference between the haemoglobin level taken in the ward and that from the blood test result with the latter being much lower. The results are tabulated in Table 1. The blood tests confirmed HIV infection and the suspicion of oral Kaposi sarcoma was thus vindicated. In consultation with the Department of Haematology of the hospital, Evans syndrome and idiopathic thrombocytopenia were added to the differential diagnosis.

The marked anaemia and thrombocytopenia on the day of admission necessitated immediate transfusion of leukocyte depleted packed red blood cells (LDPRBC) and platelets. This was done under the supervision of the attending haematologist and, on Day 1 post-admission, she received three units of LDPRBC and two mega-units of platelets. Blood tests subsequent to the transfusion revealed that her red cell count (2.92 x 1012/L), Hb (8.4 g/ dL) and platelet count (2 x 109/L) remained low. On Day 2 she received a further two units of LDPRBC and one mega-unit of platelets which resulted in her reporting feeling better. At this time, her vital signs were noted as follows: BP 109/65, heart rate 94 and temperature 36.6°C. On day 3, under the guidance of the haematologist, intravenous infusion of a 3% solution of immune globulin (Polygam®) was commenced at a rate of 1g/kg body weight over 90 minutes. As a result of the high probability of side effects with the use of Polygam®, she was also prescribed a stat dose of 100 mg of solumedrol and 25mg phenergan, administered intravenously, as prophylaxis. She received two further doses of Polygam® on Days 4 and 5. A clinical examination on Day 6 revealed that there was no intraoral bleeding, no increase in the size of the petechiae and that the melena had resolved. A further two units of LDPRBC and a mega-unit of platelets were administered due to the blood results still indicating a low haemoglobin level (6.9 g/dL) and platelet count (3 x 109/L). The lesions, however, had regressed to such a degree that the differential diagnosis of Kaposi sarcoma could then be excluded. Subsequent blood tests revealed that the direct Coombs test (direct antiglobulin test) was again positive (1+ strong micropositive), in the presence of thrombocytopenia (4 x 109/L), thus the diagnosis of Evans syndrome.
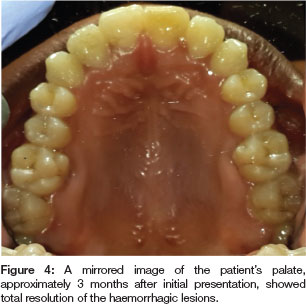
Treatment for Evans syndrome was then initiated and included: azathioprine (150 mg per os daily for three days) and prednisone (60 mg per os daily). Her red cell count (3.48 x 1012/L) and haemoglobin (10.0 g/dL) had both improved but her platelet count remained low (7 x 109/L). By Day 8 post-admission, the lesions had nearly totally regressed and her platelet count improved to 46 x 109/L. She was subsequently discharged and prednisone was prescribed (60 mg/day for one month) and the one month follow-up examination revealed total regression of all lesions. Her red cell count (3.98 x 1012/L), haemoglobin (11.5 g/dL) and platelet count (108 x 109/L) had all improved. The prednisone was continued for the next month at a dosage of 40 mg/day and, at that time, the patient's pallor and lethargy had both dissipated and she reported feeling in good health. Her blood results were within the normal range (red cell count: 3.88 x 1012/L; haemoglobin: 11.2 g/dL; platelets: 210 x 109/L). The prednisone dosage was then decreased to 35 mg/day and tapered by 5 mg/week over the following 6 weeks. Due to the very low CD4+ cell count, HAART was then commenced and at her follow up visit after a further two months, the patient had no oral or skin lesions and was otherwise stable. She is currently on a regular recall schedule and at the time of publication she remained stable without recurrence of the intra-oral haemorrhagic lesions (Figure 4).
DISCUSSION
Evans syndrome (ES) is an uncommon autoimmune disorder and is defined by the simultaneous or sequential development of autoimmune haemolytic anaemia (AIHA) and immune-induced thrombocytopenia (ITP), with a positive direct antiglobulin test (also known as the Coombs test), in the absence of known underlying aetiology. At times the condition may be accompanied by immune neutrope-nia.1,2 The condition was first described in 1951 by Robert Evans, based on a series of 24 patients who presented with a spectrum of these clinical features.1 ES is a rare condition and is diagnosed in only 0.8% to 3.7% of all patients presenting with either ITP or AIHA at onset.3 It is considered a diagnosis of exclusion and as it suggests an advanced state of immune dysregulation, other autoimmune conditions such as systemic lupus erythematosus (SLE),4 lymphoproliferative disorders,5,6 primary immuno-deficiencies7, collagen vascular diseases and autoimmune lymphoproliferative syndrome (ALPS) should be exclud-ed.8-11 In addition to the presence of ITP and AIHA seen in Evans syndrome, immunologically, there is a reversal in the CD4:CD8 ratio, 3 (normally >1) which is an indicator of severe immune dysregulation. This occurs as a result of a decrease in CD4 cells (T helper cell) with a compensatory increase in CD8 cells (a class of T regulatory cells). The reversal also occurs in uncontrolled HlV infection, however, in the case presented above only the CD4 count was available on the day of admission so no information on this ratio was available.
Haematological abnormalities such as anaemia, thrombocytopenia and neutropenia are commonly observed in patients infected with HIV.12-14 Pancytopenias are frequent complications of HIV and may be the result of a bone marrow production defect or due to increased peripheral loss or destruction of blood cells. These abnormalities may occur as a result of the HIV infection itself or as a result of HIV-associated infections or malignancies, and may also arise as a consequence of the therapy used for the HIV infection.15 Numerous studies have demonstrated that a positive Coomb's test occurs in patients with HIV, up to an incidence rate of 21%, but frank haemolysis is infrequently reported in such cases.16 HIV-associated thrombocytope-nia has also been reported based on the presence of antibodies directed against membrane proteins as well as adherence of immune complexes to platelets,16 however, ES is rarely associated with HIV infection with only a few cases having been reported.1718. To the best of our knowledge, the case presented above is unique in the sense that ES was the first presenting sign of an underlying HIV infection.
The management of Evans syndrome is challenging due to the fact that it is characterised by periods of remission and exacerbation and the response to treatment varies within the same individual.3 First-line therapy in the management of ES includes the use of corticosteroids and/ or intravenous immunoglobulin. In the acute phase, blood and/or platelet transfusions are required as symptomatic management. In the case presented in this paper the use of corticosteroids was initially avoided because of the delayed final diagnosis and the initial suspicion of Kaposi sarcoma. (Corticosteroids are contraindicated in cases of Kaposi sarcoma because of the possibility of causing an exacerbation of the lesion by inducing expression of latent Human Herpes Virus 8.)19
Second-line therapies for ES include the use of immuno-suppressive agents (cyclosporin, mycophenolate mofetil and danazol), monoclonal antibodies (rituximab) and chemotherapy (vincristine). Splenectomy may also be considered as second line therapy.3 In non-responsive cases, third-line therapy may include autologous or allo-genic stem cell transplantation but it does not have a reliable outcome, largely due to the relatively high mortality and failure rate in ES.3
CONCLUSION
The significance of oral lesions as presenting features of HIV infection and as markers for the progression of im-munosuppression is well documented. A unique case is presented above where the oral lesions associated with Evans syndrome lead to the diagnosis of a co-existing HIV infection. Normally, HIV-associated oral lesions present as opportunistic infectious lesions but, in this case, it presented as the oral lesions of a blood dyscrasia. Although other publications have reported on the co-existence of Evans Syndrome and HIV infection, as far as could be ascertained, the present case is the only one to date where Evans syndrome was the presenting condition. ES is a manifestation of severe immune dysregulation and in cases where immune-mediated destruction of blood cells are apparent, the presence of a co-existing or underlying HIV infection should be considered.
It thus begs the question: should Evans syndrome be added to the list of presenting conditions/lesions for HIV/Aids? In view of the case presented here, it certainly seems to be appropriate.
Declaration: No conflict of interest was declared.
ACRONYMS
AIHA: Autoimmune haemolytic anaemia
BP: Blood pressure
EV: Evans syndrome
Hb: Haemoglobin
HIV: Human immunodeficiency virus
INR: International normalized ratio (a measure of prothrombin time)
ITP: Immune-induced thrombocytopaenia
LDPRBC: Leukocyte depleted packed red blood cells
References
1. Evans RS, Takahashi K, Duane RT, Payne R, LiuC. Primary thrombocytopenic purpura and acquired haemolytic anemia; evidence for a common etiology. Arch Int Med 1951, 93: 341-4. [ Links ]
2. Evans RS, Duane RT. Acquired haemolytic anemia; the relation of erythrocyte antibody production to activity of the disease; the significance of thrombocytopenia and leukopenia. Blood 1949, 4: 1196-213. [ Links ]
3. Norton A, Roberts I. Management of Evans syndrome. Brit J Haematol 2005, 132: 125-37. [ Links ]
4. Deleze' M, Oria CV, Alarcon-Segovia D. Occurrence of both haemolytic anemia and thrombocytopenic purpura (Evans syndrome) in systemic lupus erythematosus. Relationship to antiphospholipid antibodies. J Rheumatol 1988, 15: 611-5. [ Links ]
5. García-Munoz R, Rodriguez-Otero P, Pegenaute C, et al. Splenic marginal zone lymphoma with Evans' syndrome, autoimmunity, and peripheral gamma/delta T cells. Ann Hematol 2009, 88: 177-8. [ Links ]
6. Hauswirth AW, Skrabs C, Schützinger C, et al. Autoimmune thrombocytopenia in non-Hodgkin's lymphomas. Haematologica 2008, 93: 447-50. [ Links ]
7. Michel M, Chanet V, Galicier L, et al. Autoimmune thrombo-cytopenic purpura and common variable immunodeficiency: analysis of 21 cases and review of literature. Medicine 2004, 83: 254-63. [ Links ]
8. Sneller MC, Straus SE, Jaffe ES, et al. A novel lymphoprolifera-tive/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest 1992, 90:334-41. [ Links ]
9. Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995, 81: 935-46. [ Links ]
10. Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in Fasassociated with human lymphoproliferative syndrome and autoimmunity. Science 1995, 268:1347-9. [ Links ]
11. Le Deist F, Emile J-F, Rieux-Laucat F, et al. Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet 1996, 348:719-23. [ Links ]
12. Harbol AW, Liesveld JL, Simpson-Haidaris PJ, Abboud CN. Mechanisms of cytopenia in human immunodeficiency virus infection. Blood Rev 1994, 8: 241-51. [ Links ]
13. Ballem PJ, Belzberg A, Devine DV, et al. Kinetic studies of the mechanism of thrombocytopenia in patients with human immunodeficiency virus infection. N Engl J Med 1992, 327:1779-84. [ Links ]
14. Baker K. The hematological complications of HIV infection. ASH education program 2003, 1: 299. [ Links ]
15. Wang W, Herrod H, Pui CH, Presbury G, Wilimas J. Immunoregulatory abnormalities in Evans syndrome. Am J Hematol 1983, 15: 381-90. [ Links ]
16. Toy PCTY, Reid ME. Positive direct antiglobulin test associated in patients with AIDS. Am J Hematol 1985, 19: 145-50. [ Links ]
17. Shashika AS, Lohiya RV. Evan's syndrome in HIV infection. J Assoc Physicians India 2012, 60:49-50. [ Links ]
18. Amid A, Leung E. Evans syndrome secondary to HIV infection. J Pediatr Hematol Oncol 2013, 35: 390-1. [ Links ]
19. Walsh CM, Nardi MA, Karpatkin S. On the mechanism of thrombocytopenic purpura in sexually active homosexual men. N Engl J Med 1984; 311: 635-9. [ Links ]
 Correspondence:
Correspondence:
wp Dreyer
PO Box 1285, Sedgefield, 6573
e-mail wpdreyer@telkomsa.net
