










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Dental Journal
On-line version ISSN 0375-1562
Print version ISSN 0011-8516
S. Afr. dent. j. vol.69 n.9 Johannesburg Oct. 2014
CASE BOOK
Oral medicine case book 64: some aspects of the pathophysiology of angioedema with special reference to the upper aerodigestive tract
M BouckaertI, II; NH WoodIII; RAG KhammissaIV; J LemmerV; L FellerVI
IBDS, PDD; Private practice, Wilgers Hospital, Pretoria, South Africa
IIBChD, MDent(MFOS), FFDRCS (Ireland); Department of Maxillofacial Oral Surgery, University of Limpopo, Medunsa Campus, South Africa
IIIBChD, PDD, MDent(OMP); Department of Periodontology and Oral Medicine, University of Limpopo, Medunsa Campus, South Africa
IVBChD, PDD, MSc(Dent), MDent(OMP); Department of Periodontology and Oral Medicine, University of Limpopo, Medunsa Campus, South Africa
VBDS, HDipDent, FCD(SA)OMP, FCMSAae, Hon.FCMSA; Department of Periodontology and Oral Medicine, University of Limpopo, Medunsa Campus, South Africa
VIDMD MDent (OMP); Department of Periodontology and Oral Medicine, University of Limpopo, Medunsa Campus, South Africa
ABSTRACT
Angioedema refers to a localized oedematous swelling of subcutaneous or sub-mucosal tissues, caused by dilatation and increased permeability of blood vessels, usually mediated either by histamine or by bradykinin.
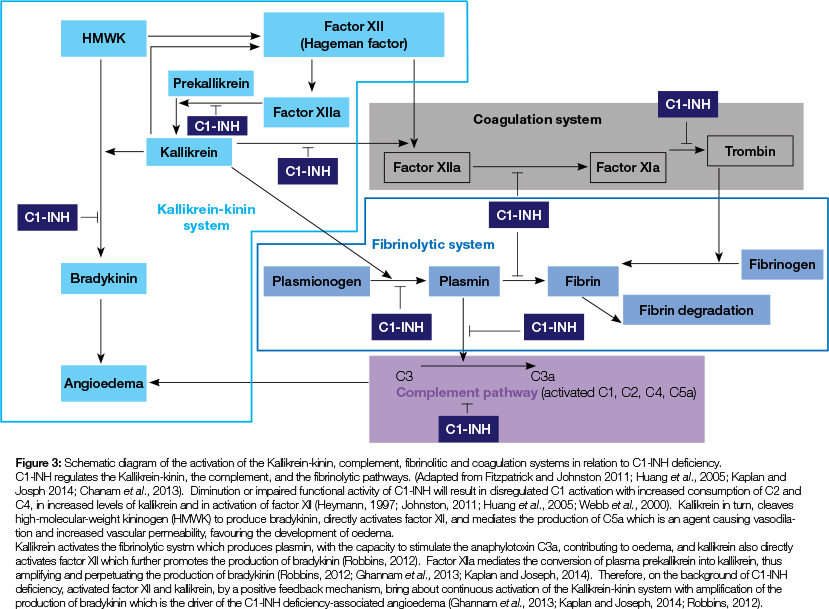
Deficiency or loss of functional activity of the complement component C1 esterase inhibitor (C1-INH) affects multiple systems, including the kallikrein-kinin, complement, coagulation and fibrinolytic pathways, and in the context of angioedema, the result is increased production and release of bradykinin and other vasoactive substances such as C3a. Owing to impairment of C1-INH, factors XIIa and kallikrein, by a positive feedback mechanism, bring about persistent activation of the kallikrein-kinin pathway with amplification of production of bradykinin, resulting in angioedema.
Histamine can cause histaminergic angioedema. As the name implies, this oedema is caused by degranulation of mast cells/ basophils as a result of an IgE-dependant allergic reaction to extracts of food, drugs, infectious agents, or to physical stimulation; or as the result of direct degranulation of mast cells/basophils independently of IgE, caused by releasing agents such as opiates, antibiotics or radio-contrast media.
As dental, oral and maxillofacial operative procedures may trigger the development of angioedema in susceptible individuals, the dental practitioner should be familiar with its signs and symptoms, its pathophysiology and with the firstline treatment of this disorder.
Key words: hereditary angioedema, bradykinin, histamine, C1-inhibitor, factor XII
ACRONYMS
ACE: angiotensin-converting enzyme
C1-INH: complement component C1 esterase inhibitor
IgE: immunoglobulin E
NSAID: non-steroidal anti-inflammatory drugs
PGE2: prostaglandin E2
INTRODUCTION
During dental, oral or maxillofacial treatment, the patient might collapse not because of the operative or surgical procedure, but because of release of vasoactive substances resulting in angioedema with upper airway obstruction. Angioedema has been defined as "localized and self-limiting oedema of the subcutaneous or submucosal tissues, owing to a temporary increase in vascular permeability caused by the release of vasoactive mediators."1 Histopathologically there is upregulation of adhesion molecules of endothelial cells with adhesion of circulating leukocytes and with a perivascular infiltrate of lymphocytes, eosinophils and neutrophils because of an increase in the endothelial intercellular spaces, and separation of perivascular collagen bundles.2,3
There are a number of variants of angioedema that may be categorized according to several possible precipitating factors and according to the nature of the specific vasoactive mediator. Angioedema may be triggered via any one of several pathogenic pathways, including firstly by histamine released from activated mast cells or basophils triggered by antigen-specific immunoglobulin E (IgE) or by mast cell degranulation precipitated by opiates, antibiotics, curare and radioactive media, independently of an IgE response.3 Secondly, angioedema may be triggered by increased levels of bradykinin as a result of either acquired or hereditary deficiency of the complement component C1 esterase inhibitor (C1 INH), or by an angiotensin-converting enzyme (ACE) inhibitor; and thirdly, in susceptible subjects, angioedema may be triggered by aspirin and other non-steroidal anti-inflammatory drugs (NSAID) against a background of altered arachidonic acid metabolism (Figure 1).3

Histamine, bradykinin and other bioactlve mediators bind to their respective receptors on endothelial cells activating intracellular pathways which bring about structural reorganization of the endothelial cell cytoskeleton, resulting in contraction of the endothelial cells with widening of the intercellular junctions,4 and leakage of a protein-rich fluid (exudate) into the extravascular tissues. The consequent increase in the osmotic pressure of the interstitial fluid, together with the increased hydrostatic pressure from increased blood flow through the dilated vessels, lead to oedema.4,5
When it occurs, this angioedema commonly affects the face (lips, cheeks and peri-orbital areas) (Figure 2) and occasionally affects the larynx, pharynx and oropharynx, and also oral tissues, particularly the tongue.2 Angioedema of the larynx, pharynx and oropharynx may be life threatening because of the risk of upper airway obstruction.6 In such cases, treatment with adrenalin, antihistamines and corticosteroids is necessary,2 and perhaps even acute emergency measures such as intubation or tracheostomy.
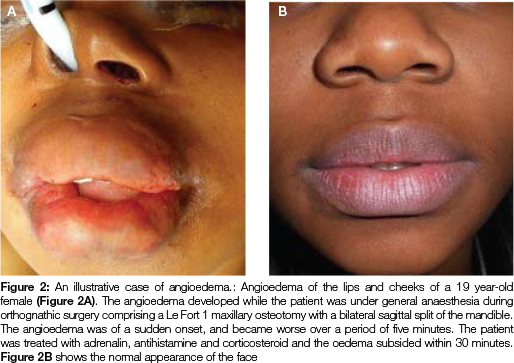
Dental treatment procedures, particularly if they are traumatic, the use of certain pharmacological agents during or after the treatment, and exposure to certain dental materials may trigger orofacial, oropharyngeal, pharyngeal or laryngeal angioedema by one of the pathways outlined above.
The purpose of this paper is to review some aspects of the pathophysiology and the management of angioedema, particularly of the upper aerodigestive tract.
HISTAMINE-MEDIATED ANGIOEDEMA
Histaminergic angioedema is rapid in onset, usually lasts less than 30 minutes and typically is accompanied by urticaria.5 IgE-mediated allergic angioedema is characterized by release of histamine, with or without other vasoactive mediators from tissue mast cells and/or blood basophils and can be precipitated by a variety of exogenous antigens such as foods, drugs, insect bites/stings, infections or by physical stimuli such as cold, heat, mechanical stimulation or exercise.1,3,5-7
Histamine interacts with H1 and H2 receptors of endothelial cells, directly causing vasodilation and increased vascular permeability, and indirectly, enhancing the local production of prostaglandins which further exaggerates the vascular responses.7-9
Anaphylaxis-like angioedema can be caused by mast cell degranulation with histamine release, independently of IgE, in response to certain drugs including opiates, antibiotics, curare or radiocontrast media;3 and by C5a and C3a components of the complement system, bringing about histamine-mediated vasodilation and increased vascular permeability.4 C5a can furthermore promote the release of cellular phospholipase A with activation of the cyclo-oxygenase pathways of arachidonic acid metabolism giving rise to PGE2, PGD2 and PGI2 which are vasodilators that significantly increase the permeability of postcapillary venules, thus playing an important role in the pathogenesis of oedema.4 The neuropeptides substance P, vasoactive intestinal peptide and somatostatin released from free nerve endings by trauma can also trigger mast cell degranulation.
Histamine-mediated angioedema develops rapidly reaching its peak within 6 hours, affects primarily the face, only rarely the larynx, and usually responds favourably to antihistamines,1,10 but clinical wisdom suggests that acute anaphy-laxis should be treated promptly first with adrenalin, followed by corticosteroids and then antihistamines.7
BRADYKININ-MEDIATED ANGIOEDEMA
Binding of bradykinin to its B1 and B2 receptors on endothelial cells may lead to their activation, with consequent dilatation and increased vascular permeability.11,12 Bradykinin can also mediate the production of nitric oxide and the mobilization of arachidonic acid metabolites, thus further promoting the inflammatory process.11,13
Bradykinin-mediated angioedema is caused by elevated plasma levels of bradykinin, is a non-histaminergic angioedema, and is not responsive to antihistamines.1,11,12 Either as a hereditary autosomal dominant trait or as an acquired impairment, in bradykinin-mediated angioedema the complement component C1-esterase inhibitor (C1-INH) may be deficient or functionally impaired. Normally, C1-INH prevents over-activation of the complement system, and is also an inhibitor of other plasma serine proteases, including components of the kallikrein-kinin system (contact system), of factor XII (Hageman factor) and factor XIa of the coagulation system and of components of the fibrinolytic system (Figure 3).5,11-18 However, there is apparently no clinical effect of C1-INH deficiency on either the coagulation or the fibrinolytic systems, so the most important role of C1-INH is to control the kallikrein-kinin and the complement pathways.16
About 85% of hereditary and acquired cases of C1-INH deficiency show diminution in C1 -INH levels (type 1), and the remainder show impaired functional activity of normal levels of C1-INH (type 2).3,17,19 Type 3 hereditary angioedema is associated with molecular alterations to the gene encoding the coagulation factor XII (Hageman factor), affects mainly women with normal plasma levels and functional activity of C1-INH, and is activated by oestrogen.5,13,14,19-21 The clinical features are identical in all the different types of hereditary angioedema.19
Hereditary C1-INH deficiency types 1 and 2 are associated with molecular variations in the gene governing C1-INH' and are characterized by low levels of C2 and C4 during episodes of angioedema' but the level of the complement protein C1q is normal.2 On the other hand, in acquired C1-INH deficiency' there are reactive auto-antibodies towards C1-INH, or a depletion of C1-INH in association with lympho-proliferative disorders or connective tissue diseases.10,16,21
Typically, acquired C1-INH is diagnosed in the fifth decade of life, and is associated with diminished levels of complement proteins C4, C2 and C1q during episodes of angioedema.7,11 Clinically, hereditary angioedema and acquired bradykinin-mediated angioedema are very similar.10
The angioedema of hereditary C1-INH deficiency is episodic, non-pruritic and non-pitting, Episodes usually start in childhood or adolescence and can affect any part of the body, but particularly the extremities, the intestines, the face and the larynx, with symptoms getting worse over 24-36 hours, and if untreated, resolving within 2-5 days.10,14 The frequency and severity of the attacks are unpredictable, but unlike in histaminergic angioedema, urticaria is seldom a feature, A family history is an important factor in establishing the diagnosis of bradykinin-mediated angioedema caused by C1-INH deficiency.10,11
The angioedematous swelling can vary from mild to severe and may even be fatal if there is obstructive laryngeal oedema, As in histaminergic angioedema, attacks may be triggered by trauma as from dental and oral surgical procedures, by physical or emotional stress, or by infection.17,18,21-23 It has been suggested that in the context of dental care, the emotional stress of dental treatment or the administration of local anaesthetic together with the associated anxiety, may trigger an angioedematous event.7 However, episodes of angioedema can occur without any identifiable precipitating factor.21
In order to reduce the frequency and the severity of the episodes, subjects with hereditary angioedema should be treated with synthetic anabolic steroids that increase the production of C1-INH by the liver, or by antifibrinolytic agents that inhibit the activation of plasmin and of factor C1.14,21 Androgens are also effective in the treatment of hereditary angioedema, but for reasons unknown they are not effective in the treatment of acquired bradykinin-mediated angioedema.10,16
For persons who are already on preventive medication for hereditary angioedema and who require dental or oral surgical treatment, it is advisable to increase the medication 5-7 days before the treatment, or alternatively to administer fresh frozen plasma or C1-INH concentrate the day before the procedure.10,14,15,21
If the patient is known to have C1-INH deficiency, than acute attacks of angioedema with severe laryngeal oedema should be treated with intravenous fresh frozen plasma or concentrate of C1-INH, together with adrenalin and perhaps with intratracheal intubation or tracheostomy.2,24 Because they only start to have an effect after some hours or even days, while anabolic steroids are necessary, they are not part of the first line of treatment.21
Since dentists and medical practitioners would not have available fresh frozen plasma or concentrate of C1-INH, subjects with a history of facial bradykinin-mediated angioedema and who need dental care should be treated in a facility with all the necessary resources.11,21
ANGIOEDEMA ASSOCIATED WITH ACE-INHIBITORS
Angiotensin-converting enzyme inhibitors which are frequently used for the treatment of hypertension or heart fail-ure,13 attenuate the degradation of bradykinin, sometimes resulting in elevated levels of bradykinin that can trigger clinical manifestations of bradykinin-mediated angioedema.1,25 Angioedema associated with ACE inhibitors usually affects the laryngeal, pharyngeal and oral mucosae, particularly the tongue,2,10,26 resulting in compromise of the airway with the potential for respiratory obstruction.25
It is estimated that between 0.1% and 6% of all persons treated with ACE inhibitors will at some time develop angioedema.5,11 When it occurs, in about 60% of cases the onset of angioedema is within a few weeks of starting the medication, but it can also occur for the first time only after several years of medication.26
If an acute emergency occurs owing to airway obstruction, as in any bradykinin-mediated angioedema, management is as previously described. Antihistamines are not effective.2,3
IDIOPATHIC ANGIOEDEMA
Recurrent angioedema in the absence of urticaria is termed idiopathic as it cannot be linked to any exogenous agent or to any underlying abnormality.2 Thus, with idiopathic angioedema, there is no family history, and the patient's serum levels, and the functional activity of C1-INH and of other components of the complement system are normal, and it may or may not respond to antihistamines.5 In severe cases, idiopathic angioedema should be treated with the standard regime of adrenalin, corticosteroids and antihistamines, though the antihistamines may or may not contribute significantly to the response.
COMMENTS
Angioedema is the result of capillary vascular dilatation and increased permeability brought about primarily by histamine-mediated allergic reactions to drugs, to extracts of food or to environmental factors; by hereditary or acquired deficiency of C1-INH (bradykinin-mediated); by ACE inhibitors (bradykinin-mediated), or by as yet unidentified factors (idiopathic).27 Most probably this classification of angioedema is an oversimplification. Although not proven, in fact, bradykinin appears to contribute to the late clinical manifestation of histaminergic angioedema.13
The problem is to clinically distinguish between the different types of acute angioedema of the upper aerodigestive tract in order to treat appropriately. Histamine-mediated anaphylactic reactions develop rapidly and must be treated promptly with adrenalin, supplemented with antihistamine and corticosteroids. Bradykinin-mediated angioedema usually develops insidiously over a few hours and should ideally be treated with C1-INH concentrate, or with fresh frozen plasma intravenously, but these agents are not readily available. Unfortunately because these reactions develop gradually, they usually occur after the patient has left the health care facility or the doctor's rooms, and are thus seldom recognized or treated appropriately.
References
1. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy 2014;69:602-16. [ Links ]
2. Kaplan AP Urticaria and Angioedema. In: Wolff K, Goldsmith L, Katz S, Gilchrest B, AS P, Leffell D, editors. Fitzpatrick's Dermatology in General Medicine. 7 ed. New York: McGraw-Hill; 2008: 330-42. [ Links ]
3. Edwards J. Urticaria and Angioedema. In: Kasper D, Hauser S, Longo D, Jameson J, Loscaizo J, editors. Harrison's Principles of Internal Medicine. 17 ed. New York: McGraw-Hill; 2008. 1951-4. [ Links ]
4. Kumar V, Abbas AK, Fausto N. Acute and chronic inflammation. In: Kumar V, Abbas AK, Fausto N, editors. Pathologic Basis of Disease. Seventh Edition ed: Elsevier Saunders; 2005:47 - 86. [ Links ]
5. Bork K. Angioedema. Immunol Allergy Clin North Am 2014;34:23-31. [ Links ]
6. Ogasawara T, Kitagawa Y, Ogawa T, Yamada T, Kawamura Y, Sano K. MR imaging and thermography of facial angioedema: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001;92:473-6. [ Links ]
7. Rees SR, Gibson J. Angioedema and swellings of the orofacial region. Oral Dis 1997;3:39-42. [ Links ]
8. Wasserman SI. Biochemical mediators of allergic reactions. In: Patterson R, Zeiss CR, Grammer LC, Greenberger PA, editors. Allergic Diseases, Diagnosis and Management. Fourth edition. Philadelphia, USA: J.B. Lippincott Company; 1993: 57 - 72. [ Links ]
9. Metzger WJ. Urticaria, angioedema, and hereditary angioedema. In: Patterson R, Zeiss CR, Grammer LC, Greenberger PA, editors. Allergic Diseases, Diagnosis and Managment. Fourth edition. Philadelphia, USA: J.B. Lippincott Company; 1993: 331 - 52. [ Links ]
10. Cicardi M, Zanichelli A. Diagnosing angioedema. Immunol Allergy Clin North Am 2013;33:449-56. [ Links ]
11. Douglas T, Johnston DO. Diagnosis and management of hereditary angioedema. J Am Osteopath Assoc 2011 ; 111:28-36. [ Links ]
12. Bossi F, Tedesco F. Role of B1 bradykinin receptor and gC1qR/p33 in angioedema. Immunol Allergy Clin North Am 2013;33:535-44. [ Links ]
13. Kaplan AP, Joseph K. Pathogenic mechanisms of bradykinin mediated diseases: dysregulation of an innate inflammatory pathway. Adv Immunol 2014;121:41-89. [ Links ]
14. Huang YT, Lin YZ, Wu HL, Chiu TF, Lee KM, Tsai HY, et al Hereditary angioedema: a family study. Asian Pac J Allergy Immunol 2005;23:227-33. [ Links ]
15. Webb MD, Hakimeh S, Holly LK. Management of children with hereditary angioedema: a report of two cases. Pediatr Dent 2000;22:141-3. [ Links ]
16. Heymann WR. Acquired angioedema. J Am Acad Dermatol 1997;36:611-5. [ Links ]
17. Cifuentes J, Palisson F, Valladares S, Jerez D. Life-threatening complications following orthognathic surgery in a patient with undiagnosed hereditary angioedema. J Oral Maxillofac Surg 2013;71:e185-8. [ Links ]
18. Van Sickels NJ, Hunsaker RB, Van Sickels JE. Hereditary angioedema: treatment, management, and precautions in patients presenting for dental care. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;109:168-72. [ Links ]
19. Scully C, Langdon J, Evans J. Marathon of eponyms: 17 Quincke oedema (Angioedema). Oral Dis 2011;17:342-4. [ Links ]
20. Bork K. Hereditary angioedema with normal C1 inhibitor. Immunol Allergy Clin North Am 2013;33:457-70. [ Links ]
21. Morcavallo PS, Rossi G, Martini M, Carini F. Hereditary angioedema in oral surgery: Overview of the clinical picture and report of a case. J Oral Maxillofac Surg 2010;68:2307-11. [ Links ]
22. Bork K, Hardt J, Staubach-Renz P, Witzke G. Risk of laryngeal edema and facial swellings after tooth extraction in patients with hereditary angioedema with and without prophylaxis with C1 inhibitor concentrate: a retrospective study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2011;112:58-64. [ Links ]
23. Puricelli E, Ponzoni D, Artuzi FE, Martins GL, Calcagnotto T. Clinical management of angioneurotic oedema patient post-orthognathic surgery. Int J Oral Maxillofac Surg 2011;40:106-9. [ Links ]
24. Rice S, Cochrane TJ, Millwaters M, Ali NT. Emergency management of upper airway angio-oedema after routine dental extraction in a patient with C1 esterase deficiency. Br J Oral Maxillofac Surg 2008;46:394-6. [ Links ]
25. Wilson M, Frohna W, Trent G, Sauter D. Evaluating for seasonal variation in angiotensin-converting enzyme inhibitor- and angiotensin receptor blocker-induced angioedema. Ann Allergy Asthma Immunol 2014;112:178-9. [ Links ]
26. Awoniyi CA, Yannaras S, Bauerfeind JM. Acute angioedema in a patient on long-term angiotensin converting enzyme inhibitor following oral surgery: A case report. Open Anesthesiol J 2013;7:49-51. [ Links ]
27. Chan YF, Kalira D, Hore P. Angiotensin-converting enzyme inhibitors as a cause of unilateral tongue angioedema in a 68-year-old woman. Am J Emerg Med 2006;24:249-50. [ Links ]
 Correspondence:
Correspondence:
L Feller
Head: Dept. Periodontology and Oral Medicine
Box D26 School of Dentistry
MEDUNSA 0204
South Africa
Tel: (012) 521 4834
Email: liviu.feller@ul.ac.za

{kind=link}