










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.103 n.12 Pretoria Dec. 2013
MOLECULAR GENETICS
Genetic testing for Duchenne/Becker muscular dystrophy in Johannesburg, South Africa
R KerrI; C RobinsonII; F B EssopIII; A KrauseIV
IPhD, Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIBSc (Hons), Division of Human Genetics, National Health Laboratory Service, Johannesburg, South Africa
IIIMSc (Med), Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, Division of Human Genetics, National Health Laboratory Service, Johannesburg, South Africa
IVMB BCh, PhD, Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, Division of Human Genetics, National Health Laboratory Service, Johannesburg, South Africa
ABSTRACT
BACKGROUND: Genetic testing for Duchenne/Becker muscular dystrophy (DMD/BMD) mutations initially involved multiplex polymerase chain reaction (mPCR), which targeted two mutation hotspots in the gene and detected deletions in affected males. A newer technology, multiplex ligation-dependent probe amplification (MLPA), was introduced for diagnostic testing in 2007.
OBJECTIVES: To evaluate MLPA relative to mPCR as a technique for DMD/BMD diagnostic testing and to establish whether the mutation profile in affected individuals differs between different South African ethnic groups.
METHODS: From January 2000 - May 2007, genetic diagnostic testing for DMD/BMD was undertaken in 128 male patients using mPCR. From May 2007 onwards, MLPA replaced this technique and 261 males were investigated. MLPA is a kit-based technology available from MRC-Holland.
RESULTS: Of the 128 and 261 probands tested using mPCR and MLPA, respectively, 31% and 34% were found to carry a deletion mutation. Further, MLPA could detect duplication mutations (11.5%), complex rearrangements (1.5%) and small mutations (1.5%). In black patients, deletion mutations were found to cluster in the 3' region of the gene. No population-specific pathogenic mutations were found.
CONCLUSIONS: The mutation detection rate for mPCR and MLPA is similar for deletion mutations, but MLPA proved to be a better diagnostic approach as it could detect other types of mutations as well, including duplications, complex rearrangements and small mutations. MLPA could also diagnose mutation status in at-risk female relatives, which is not possible with mPCR.
Duchenne muscular dystrophy (DMD) (OMIM #310200) is an X-linked recessive inherited muscle-wasting disease characterised primarily by progressive weakness and atrophy of the skeletal and cardiac muscle. DMD patients are typically wheelchair-bound by 12 years of age and die in their late teens or early twenties of respiratory failure. It is one of the most common Mendelian disorders, occurring in all population groups with a birth prevalence of approximately 1/3 500 males. The disease gene, DMD, linked to chromosome Xp21.2, was one of the first genes to be isolated by positional cloning,[1] and the deficient product in affected boys was then identified as the protein dystrophin.[2] Dystrophin localises to the cell membrane in muscle cells and binds the protein actin, in this way forming part of the protein complex which links the cytoskeleton with the cell membrane. The gene is extremely large, spanning 2 400 kb of genomic DNA and comprising 79 exons which encode a 14 kb transcript. Causative mutations are loss-of-function mutations:[3] approximately 65% are out-of-frame deletions of one or more complete exons; 5 - 10% are duplications; and the remainder are nonsense, splice site or frameshift mutations; missense mutations are rare. Complex rearrangements are seen in ~2% of DMD cases. The new mutation rate is high, explained in part by the large size of the gene, and in approximately 1/3 of cases, the mother of an affected boy is not a carrier. Becker muscular dystrophy (BMD) (OMIM #300376) is a milder form of the disease, caused by mutations in the same gene. The effect of the mutations is different for BMD, and they lead to either a reduction in the amount, or an alteration in the size of the dystrophin protein, but not to a complete loss of protein as in DMD.[4] The mutation profile in BMD is different to DMD,[3] with ~85% of BMD mutations being accounted for by in-frame deletions, 5 - 10% by duplication mutations and the remainder by point mutations, splice site or small insertions/deletions.
The 5' proximal end of dystrophin is more susceptible to duplication events and the most common exonic duplication mutation in DMD/ BMD patients has been shown to involve exon 2.[5] DMD and BMD are essentially clinically similar disorders, falling under the umbrella classification of 'dystrophinopathies', with DMD being the severe end of the spectrum and BMD a milder phenotype. The 'reading frame rule' may be applied to predict severity of disease in young children who represent simplex cases - if the mutation (deletion or duplication) results in an out-of-frame mutation, then this usually correlates with a more severe phenotype (DMD), and in-frame mutations generally correlate with the milder BMD phenotype.[4]
Two recombination hotspots for the large deletions have been identified: the proximal hotspot spans exons 2 - 7[6] and the distal hotspot spans exons 45 - 51.[7] Several multiplex polymerase chain reaction (mPCR) approaches have been developed to test for these deletions (testing approaches and PCR primer sequences can be found at the Leiden Open Variation Database (LOVD): http://www. lovd.nl/dmd). These work on the principle that failure of an exon to amplify by PCR in hemizygous males indicates that the exon is deleted. PCR is a qualitative rather than a quantitative assay and is therefore insensitive to the copy number of the target sequence. Thus, duplication mutations in affected boys, and duplication or deletion mutations in heterozygous females cannot be identified using this technique. Consequently, carrier testing in at-risk females is not possible using the mPCR technique, and - prior to multiplex ligation- dependent probe amplification (MLPA) - was therefore carried out in the Molecular Diagnostic Laboratory, Division of Human Genetics, South African Institute for Medical Research (SAIMR) (now, the National Health Laboratory Service (NHLS)), University of the Witwatersrand, Johannesburg, by performing linked marker analysis.
MLPA is a relatively simple, high-throughput and robust method developed by MRC-Holland.[8] The technique is based on the semi-quantitative PCR principle and can be applied for detecting copy number changes. Using MLPA, the diagnosis of DMD/BMD-causing mutations has become more comprehensive as probes have been designed to detect deletions and duplications of all 79 exons of the dystrophin gene. This allows for accurate diagnostic, carrier and prenatal testing. MLPA can also identify smaller mutations if they occur within the probe-binding regions.[9] MLPA is currently the technique of choice as a first-line screen to detect deletions/ duplications in routine DMD/BMD genetic diagnostics.[10]
There are few published reports on DMD mutations in South African populations. Previous studies in this country have identified deletions in 50% (30/60) of affected boys, using the Southern blotting technique,[11] 42% (46/110) of affected boys using Southern blotting together with mPCR,[12] and 57% (39/68) of affected boys using the
Chamberlain and Beggs mPCR approach.[13] All these studies included patients of different ethnic origins (black, Indian, mixed ancestry and white). Ballo et al.[12] reported a lower incidence of deletions in black patients (10/42; 26%) and suggested that other intragenic mutations, lying outside the two described hotspots, may be causative in this population group. However, Hallwirth Pillay et al.[13] did not find the same picture in their cohort of black patients, reporting a deletion detection rate similar to that in the literature (25/40; 62%).
Objectives
To (i) evaluate MLPA as a technique used for DMD/BMD diagnostic testing compared with the mPCR used previously in the laboratory; (ii) review the results of the testing performed in the laboratory to date; and (iii) assess and review the mutation profile in a larger cohort of DMD/BMD South African patients. The latter is important to characterise in light of new pharmacogenetic, or other therapeutic approaches which target specific mutations or gene regions.
Methods
Although a genetic counselling service for affected families has been available locally for four decades, a molecular diagnostic service for DMD/BMD was only established at the Division of Human Genetics, SAIMR and University of the Witwatersrand, in 2000. Prior to this, samples were referred for testing to the Department of Human Genetics at the University of Cape Town, where molecular testing techniques had been established and were available.
Subjects
Patients were referred for DMD/BMD diagnostic testing from medical geneticists and genetic counsellors from the Clinical Section, Division of Human Genetics, and from doctors in public and private hospitals in the areas surrounding Johannesburg. Testing requests were also received from neighbouring provinces (Limpopo and KwaZulu-Natal). From January 2000 to May 2007, 128 unrelated probands were referred for such testing. These patients were investigated using an in-house mPCR method. During this time period, 45 carrier tests were undertaken in female family members, using linked marker analysis. Patients referred from May 2007 to April 2013 were tested using MLPA analysis. MLPA was performed on 261 probands and 57 at-risk female family members. In total, from April 2000 to April 2013, 11 prenatal tests have been carried out. Controls for all techniques were from unaffected, unrelated individuals available in the laboratory.
DNA extraction and quantification
For molecular analysis on probands and at-risk female family members requesting testing, blood was collected in EDTA tubes and DNA was extracted using either the salting-out protocol[14] or the High Preparation PCR Extraction kit (Roche Diagnostics) for samples where <1 ml of blood was received. DNA was quantified using a nanodrop spectrophotometer. Prenatal testing, following gender determination, was performed on uncultured samples obtained either by chorionic villus sampling or amniocentesis. DNA was extracted from prenatal samples using a phenol-chloroform protocol.[15]
mPCR assay and electrophoresis
mPCR was designed using the primer sequences for exons 4, 5, 8, 13, 17, 19, 30, 42 - 45, 47, 49, 51, 60 and 62 described by Yau et al.[16] Primers for exons 1, 7, 9, 12 and 52 were obtained from the LOVD for DMD. Primers were fluorescently labelled. Each PCR run included a positive and a negative control sample. mPCR was performed using 100 - 200 ng of DNA. PCR products were loaded on the ABI377 Genetic Analyzer or on the ABI 3130xl Genetic Analyzer (Applied Biosystems). The presence/ absence of a PCR product was determined using GeneScan (ABI377) or GeneMapper version 4.0 (ABI 3130xl) software.
MLPA analysis
MLPA analysis was performed using two SALSA MLPA kits specific for DMD/BMD testing (MRC-Holland). The SALSA P034 kit contains probes for exons 1 - 10, 21 - 30, 41 - 50 and 61 - 70 of the dystrophin gene while the SALSA P035 kit contains probes for the other exons in the dystrophin gene: exons 11 - 20, 31 - 40, 51 - 60 and 71 - 79. Each MLPA run included four normal female control samples, one deletion-positive control sample and one duplication-positive control sample. MLPA was performed with 5 µl of DNA (diluted in Tris-EDTA buffer to 40 ng/µl, according to the manufacturer's instructions. PCR products were separated by electrophoresis on the ABI 3130xl Genetic Analyzer. The relative peak heights were extracted from the ABI 3130xl Genetic Analyzer using GeneMapper version 4.0 and analysed using an MS Excel-based programme developed by the National Genetics Reference Laboratory, Manchester, UK (http://www.ngrl.org.uk/Manchester/projects/informatics/mlpa).
Sequencing analysis
Single exon deletions identified by MLPA were confirmed by standard PCR using primers from the mPCR (if the exon deleted was included in the mPCR multiplexes) or by ordering new primers with alternative primer binding sites. These PCR products were visualised on a 3% agarose gel (Whitehead Scientific). If the deleted exon did not amplify, this confirmed the deletion. If the deleted exon was present in the second assay, the exon was sequenced using automated Sanger sequencing.[17] Following sequencing, the samples were run with 10 µl Hi-Di formamide (Applied Biosystems) on the ABI 3130xl Genetic Analyzer. The results were analysed using Sequencing Analysis (Applied Biosystems) and Laser Gene, DNA Star software.
Linked marker analysis
Families where the proband tested negative for deletions using mPCR and negative for deletions/duplications using MLPA were also offered linked marker analysis to identify the high-risk X chromosome. Carrier testing by linked marker analysis was performed using six intragenic, fluorescently labelled markers: 5'DYS1, 5'DYS7, 5'DYS5, IVS44A, STR49 and DXS1214. Primer sequences for these linked markers were obtained from the LOVD website. Each PCR run included the affected proband in the family and a normal female and a normal male control sample. The six linked markers were divided into two multiplex reactions (IVS44A, 5'DYS5, DXS1214 and 5'DYS7, 5'DYS1, STR49). PCR products were loaded on the ABI377 Genetic Analyzer or on the ABI 3130xl Genetic Analyzer. The relative peak sizes were analysed using GeneScan (ABI377) or GeneMapper version 4.0 (ABI 3130xl) software to resolve dinucleotide repeat number.
Ethics consent for this study was obtained from the Ethics Committee of the University of the Witwatersrand (clearance certificate M110937).
Results
mPCR
Among the 128 probands tested using mPCR, 40 (31%) deletion mutations were identified (Table 1).

MLPA
Contiguous exon deletion or duplication results, as well as complex rearrangements were considered true findings and were not pursued further. Single exon deletion results on both MLPA and mPCR needed to be investigated to identify true single exon deletions and exclude the possibility of the presence of either a benign probe/ primer-binding mutation or a pathogenic probe/primer-binding mutation, both of which would give a false-positive 'deletion' mutation result.
Mutations were detected in 128/261 (49%) probands tested using MLPA (Table 2). The detection rate for each ethnic group was as follows: blacks 47.5%, Indians 53% and whites 42.5%.
Mutation profiles
The 128 mutations identified through MLPA analysis were separated into different categories for further consideration: 85 (66%) were contiguous exon deletions, 23 (18%) were contiguous exon duplications, five (4%) were true single exon deletions, seven (6%) were single exon duplications, four (3%) were complex rearrangements and four (3%) were small mutations. The analysis of deletion or duplication mutations by position and ethnic background is provided in Table 3 and the complex rearrangements and small mutations are described in further detail below.
The majority of the contiguous exon deletion mutations (in all ethnic groups) were unique, except for a deletion of exon 3 - 7, which occurred in 4/63 black patients and in 3/17 of white patients (it was not determined if the breakpoints were similar). All contiguous duplication mutations were unique, except for a duplication of exons 14 - 17, which occurred in 2/19 black patients. In black patients who carried a contiguous exon deletion mutation (n=63), the mutation was found in the 3' distal region of the gene in 44 (70%) patients, showing that it is 2.3 times more likely for deletion mutations to occur in the distal region of the gene in this population group. The clustering of deletion mutations in the 3' deletion hotspot occurs globally,[3] so this result is in keeping with previously reported observations. The sample size of the Indian and white cohorts was small, and this possibly explains why this pattern was not observed in these ethnic groups. Contiguous and single exon duplication mutations were found more commonly in the 5' proximal region of the gene (in all ethnic groups), this being in keeping with the international literature.[3]
MLPA single exon deletions
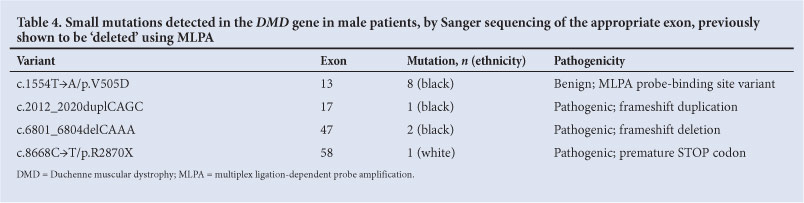
If the MLPA result was corroborated using PCR, a single exon deletion was confirmed in the patient. If PCR produced an amplification product, then the product was Sanger sequenced. Small mutations detected following sequencing are summarised in Table 4.
Complex rearrangements
Four probands were found to have unusual MLPA results. Three of these patients had non-contiguous exon duplications (an example is given in Fig. 1A) and one patient was found to have a contiguous exon deletion and a 7-fold non-contiguous exon duplication suggesting a complex rearrangement (Fig. 1B).
Prenatal testing
Before 2007 (prior to the availability of MLPA technology), fetal sexing was performed initially, followed by testing for the family mutation in a male fetus. From 2007 onwards, the presence of the family mutation could be tested using MLPA. Irrespective of time period, if the family mutation had not been identified, inheritance of the high-risk X chromosome from the mother was tracked in a male fetus using linked marker analysis. From 2000 to 2012, 11 prenatal tests were undertaken. The family mutation was identified and screened for in 10/11 cases and one case was worked up using linked markers. The fetus was female in only one case (a reflection of the approach whereby ultrasound is used to sex the fetus and invasive prenatal testing is only performed on male fetuses or where fetal sex cannot be determined). In the 10 cases where the fetus was found to be male, a negative result for the familial pathogenic mutation was found in six cases, and four cases were positive for the familial mutation.
Alternate diagnoses
In cases where the clinical picture was unclear, testing requests for additional muscular dystrophies might have been requested by the referring clinician. These included requests for Charcot-Marie-Tooth type 1A, myotonic dystrophy and spinal muscular atrophy (SMA). A positive SMA result was found in five of the probands in this cohort. Further, in white patients, limb-girdle muscular dystrophy type 2I may present with a BMD/DMD phenotype (manuscript in preparation).
Discussion
In a multi-ethnic South African cohort of 390 DMD/BMD probands tested for DMD mutation status, it was found that MLPA has a higher detection rate than mPCR (49% v. 31%, respectively), which is consistent with the international literature. Considering that the overall detection rate for deletion mutations is similar for MLPA (34%) and mPCR (31%), it is clear that the advantage of MLPA lies mostly in the fact that it can detect duplication mutations (a further 11.5% of mutations detected) and complex rearrangements (a further 1.5% of mutations detected), and further deletions in regions not covered by mPCR. Small mutations that occur within MLPA probe-binding sites were also found (another 1.5% of mutations detected). This is an unintended use of MLPA but presumably also occurs with mPCR. The mutation profile for the different ethnic groups studied here (black, Indian and white South African) is similar, with inconsistencies possibly being attributable to small sample size. This study does verify, however, that distal deletions in blacks are more common, and the distribution of deletion mutations in Indians and white South Africans should be confirmed on a larger cohort. The position of a deletion may be important because the exon-skipping gene therapy approach is aimed at distal mutations.
Since 33% of affected boys and 33% of mothers carry de novo mutations,[18] the lack of population-specific mutation profiles is not unexpected. Overall, deletion mutations were 3 - 4 times more common than duplications in all ethnic groups, and complex rearrangements and small mutations were rare.
While the reported detection rate for deletion mutations in the international literature is over 60%, in our laboratory lower detection rates have been found, irrespective of the molecular technique used for testing. It is suggested that this is a reflection of lack of rigour on the part of the clinicians referring a patient for DMD molecular testing, and not a reflection on the mutation profile of local DMD/ BMD patients. In certain patients, multiple genetic tests were requested, showing that DMD testing is sometimes being requested to exclude the diagnosis. Following from this study, it is apparent that more clearly defined clinical parameters need to be met before genetic testing is undertaken, including elevated serum creatine kinase levels.
This study shows that using MLPA analysis for diagnostic, prenatal and carrier testing increases the sensitivity of identifying both deletion and duplication mutations in all 79 exons of the DMD gene. MLPA also detects complex rearrangements, although it does not define break points of duplicated material within the gene, which may be relevant for gene therapy. While sequencing analysis was used to confirm single exon deletions, ideally sequencing should also be used to confirm the extent of multi-exon deletion or duplication mutations.
It is very important to be aware of population-specific variants that occur within the MLPA probe-binding regions as these can lead to false-positive results and misdiagnosis. When undertaking MLPA, it is imperative to establish an understanding of the local population variation. In the South African black population, a missense variant (c.1554T→A/p.V505D) was found in the probe-binding region of exon 13. This variant is non-pathogenic, but gives a false-positive deletion result on MLPA analysis and occurs at a significant frequency.
MLPA is a direct and accurate method of testing that is now offered to at-risk females for carrier testing. The technique was used to establish the carrier status in 57 female relatives, negating the need for a lengthy and expensive linked-marker family work-up. MLPA is especially useful in sporadic/new mutation cases, where linked markers could refine risk but are not definitive, which mutation testing is. As MLPA is a direct mutation detection approach, it is also useful in prenatal testing cases where mothers may be germline mosaics.
Conclusion
The need to identify the pathogenic mutation in individuals affected by DMD is more apparent than ever, as significant progress is being made on the therapeutic front. The most promising therapeutic approaches are nonsense mutation read-through and antisense oligonucleotide-induced exon skipping.[19] While nonsense mutations cause about 10% of DMD, theoretically over 80% of patients could be treated with antisense oligonucleotides. By this approach, the cellular machinery can be 'fooled' into bypassing an exon containing a point mutation/deletion/duplication such that the dystrophin protein is still produced, even though it will be in an altered form - with the net clinical effect of converting DMD to a BMD phenotype. Early diagnosis, identification of the mutation and intervention will be key to any therapeutic approach. The finding that deletion mutations do cluster in the 3' distal region of the gene in black patients is of significance when considering therapeutic approaches.
Acknowledgements
We thank members of the Molecular Genetic Diagnostic Laboratory, Division of Human Genetics, NHLS, particularly Karen Kuhn and Elana Vorster, for laboratory support.
References
1. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987;31;50(3):509-517. [http://dx.doi.org/10.1016/0092-8674(87)90504-6] [ Links ]
2. Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987;24;51(6):919-928. [http://dx.doi.org/10.1016/0092-8674(87)90579-4] [ Links ]
3. Darras BT, Miller DT, Urion DK. Dystrophinopathies. In: Pagon RA, Adam MP, Bird TD, et al, eds. GeneReviews. Seattle: University of Washington, 1993-2013. http://www.ncbi.nlm.nih.gov/books/NBK1119/ (accessed 10 October 2013). [ Links ]
4. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2(1):90-95. [http://dx.doi.org/10.1016/0888-7543(88)90113-9] [ Links ]
5. White SJ, Aartsma-Rus A, Flanigan KM, et al. Duplications in the DMD Gene. Hum Mutat 2006;27(9):938-945. [http://dx.doi.org/10.1002/humu.20367] [ Links ]
6. Liechti-Gallati S, Koenig M, Kunkel LM, et al. Molecular deletion patterns in Duchenne and Becker type muscular dystrophy. Hum Genet 1989;81(4):343-348. [http://dx.doi.org/10.1007/BF00283688] [ Links ]
7. Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990;86(1):45-48. [http://dx.doi.org/10.1007/BF00205170] [ Links ]
8. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30(12):e57. [http://dx.doi.org/10.1093/nar/gnf056] [ Links ]
9. Okizuka Y, Takeshima Y, Awano H, Zhang Z, Yagi M, Matsuo M. Small mutations detected by multiplex ligation-dependent probe amplification of the dystrophin gene. Genet Test Mol Biomarkers 2009;3(3):427-431. [http://dx.doi.org/10.1089/gtmb.2009.0002] [ Links ]
10. Abbs S, Tuffery-Giraud S, Bakker E, Ferlini A, Sejersen T, Mueller CR. Best practice guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscular Disord 2010;20(6):422-427. [http://dx.doi.org/10.1016/j.nmd.2010.04.005] [ Links ]
11. Ballo R, Hitzeroth HW, Beighton PH. Duchenne muscular dystrophy - a molecular service. S Afr Med J 1991;79(4):209-212. [ Links ]
12. Ballo R, Viljoen D, Beighton PH. Duchenne and Becker muscular dystrophy prevalence in South Africa and molecular findings in 128 persons affected. S Afr Med J 1994;84(8 Pt 1):494-497. [ Links ]
13. Hallwirth Pillay KD, Bill PLA, Madurai S, Mubaiwa L, Rapiti P. Molecular deletion patterns in Duchenne and Becker muscular dystrophy patients from KwaZulu Natal. J Neurol Sci 2007;252(1):1-3. [http://dx.doi.org/10.1016/j.jns.2006.06.025] [ Links ]
14. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16(3):1215. [http://dx.doi.org/10.1093/nar/16.3.1215] [ Links ]
15. Barker, K. At the Bench: A Laboratory Navigator. New York; Cold Spring Harbor Press, 1998. [ Links ]
16. Yau SC, Bobrow M, Mathew CG, Abbs SJ. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet 1996;33(7):550-558. [http://dx.doi.org/10.1136/jmg.33.7.550] [ Links ]
17. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci 1977;74(12):5463-5467. [http://dx.doi.org/10.1073/pnas.74.12.5463] [ Links ]
18. Edwards J. The population genetics of Duchenne: Natural and artificial selection in Duchenne muscular dystrophy. J Med Genet 1986;23(6):521-530. [http://dx.doi.org/10.1136/jmg.23.6.521] [ Links ]
19. Laing NG, Davis MR, Bayley K, Fletcher S, Wilton SD. Molecular diagnosis of Duchenne muscular dystrophy: Past, present and future in relation to implementing therapies. Clin Biochem Rev 2011;32(3):129-134. [ Links ]
 Correspondence:
Correspondence:
R Kerr
(robyn.kerr@nhls.ac.za)
Accepted 12 August 2013.

{kind=link}
{kind=link}
{kind=link}