










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489Print version ISSN 0038-2353
S. Afr. j. sci. vol.105 n.7-8 Pretoria Jul./Aug. 2009
RESEARCH LETTERS
Photocatalytic degradation of methyl red dye
M.A. MahmoudI, *; A. PoncheriII; Y. BadrIII; M.G. Abd El WahedI
IChemistry Department, Faculty of Science, Zagazig University, P.O. Box 44519, Zagazig, Egypt
IILaser Dynamics Laboratory, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332-0400, U.S.A.
IIINational Institute of Laser Enhanced Science, Cairo University, Cairo, Egypt
ABSTRACT
Silica nanoparticles (SiO2 NPs) are active in the photocatalytic degradation of methyl red dye (MR). SiO2 NPs and SiO2 NPs that have been doped with either silver (Ag NPs) and/or gold nanoparticles (Au NPs) were prepared. The particle size and morphology of the catalysts were assessed by transmission electron microscopy (TEM) imaging. The rate of photocatalytic degradation of MR was found to increase from SiO2 NPs, SiO2 NPs coated with both Au NPs and Ag NPs, SiO2 NPs coated with Ag NPs, SiO2 NPs coated with Au NPs, Ag+-doped SiO2 NPs to Au3+-doped SiO2 NPs.
Key words: silica nanoparticles, photocatalysis, photodegradation, gold, silver
Introduction
Most dyes used in the pigmentation of textiles, paper, leather, ceramics, cosmetics, inks and food-processing products are derived from azo dyes, which are characterised by the presence of one or more azo groups (-N=N-) in their structure.1 Approximately 15% of the dyes produced worldwide are lost within waste water during synthesis and processing.2 This waste represents a great hazard to human and environmental health due to the toxicity of azo dyes.3
Although silica is essentially inert for many reactions, it shows noticeable activity towards certain catalytic4–6 and photocatalytic7–11 reactions. Pure silica was proven to promote photocatalytic reactions such as the photo-oxidation of COref. 13 and the photometathesis of propene.13,14 Additionally, silica-based photocatalysts, such as silica-alumina,15,16 silica-supported zirconia,7,17 silica-supported magnesia,8 and silica-alumina-titania,9,18 exhibit activity under UV irradiation at room temperature.
The photoactive sites were formed on the surface of silica (SiO2) prepared by a sol-gel method.19 These active sites were revealed by vacuum (V)UV-UV, infrared (IR), electron spin resonance (ESR) and photoluminescence spectroscopy.20 The IR symmetric stretching vibration of the Si-O– non-bridging bond appeared at 950 cm–1. Furthermore, the enhancement of the peak intensity at 950 cm–1 indicated an increase in the amount of the non-bridging oxygen, i.e. a change in the structural units from SiO2 (three-dimensional network structure) to SiO4 (isolated tetrahedron).21 The silica nanoparticles could be photoexcited under UV light (258 nm), which corresponded to a charge transfer from the bonding orbital of Si–O to the 2p non-bonding orbital of non-bridging oxygen.22
There are conflicting reports in the literature describing the effects of metal ions in the presence of semiconductor photocatalysts. In some cases they may act as a rate accelerator23,24 but, in other cases, the rate was found to decrease upon the addition of such metal ions.25,26 In this study, we prepared silica nanoparticles and modified the surface by doping with metal ions or coating with metallic nanoparticles. We also studied the effect of additives on the kinetics and the mechanism of photocatalytic degradation of the MR dye.
Methods
Methyl red (MR), ethanol, hydrogen tetrachloroaurate (HAuCl4) and tetraethylorthosilicate (TEOS) were purchased from Sigma-Aldrich; while silver nitrate (AgNO3), nitric acid (HNO3) and sodium borohydride (NaBH4) were obtained from Fluka.
Preparation of silica nanoparticles (SiO2 NPs)
The SiO2 NPs were prepared from TEOS by HNO3-catalysed hydrolysis27. Tetraethylorthosilicate and ethanol, in volumes of 10 ml, were mixed together; the resulting mixture was added drop-wise to 60 ml of a 0.1 M HNO3 solution. The final mixture was stirred for 8 h at 60°C for complete hydrolysis of TEOS to SiO2 NPs. The concentration of the SiO2 NPs solution, after it was diluted to 90 ml by distilled water, was found to be 30 g l–1. These particles were centrifuged, washed with ethanol and dried in air. The particles were then used for coating for catalysis without further treatment.
Preparation of SiO2 NPs doped with Ag+ or Au3+
SiO2 NPs doped with 0.1% Ag+ or Au3+ ions were prepared by thoroughly mixing 1 mM of AgNO3 or HAuCl4 with 100 ml of 0.999 M SiO2 NPs in doubly-distilled water. The solution was then allowed to stand for 2 h to allow the ions to be adsorbed completely.28 The resulting particles were placed under vacuum to facilitate complete dryness.
Preparation of metal NPs on the surface of SiO2 NPs
Ag NPs, Au NPs, or co-deposited NPs were adsorbed to the surface of SiO2 NPs with a molecular ratio of 0.1%. Deposition of pure metal samples was accomplished by mixing 1 mM of AgNO3 (or HAuCl4) with 80 ml of SiO2 NPs (0.999 M) under 5 min of vigorous stirring. Next, 1 mM NaBH4 was added and the solution was diluted to 100 ml with doubly-distilled water29. To prepare Au NPs and Ag NPs co-deposited on the surface of SiO2 NPs, 0.5 mM of AgNO3 was mixed with 0.999 M and the solution was stirred for 5 min. Then 0.5 mM NaBH4 was added. Thereafter, 0.5 mM of HAuCl4 was added, followed by an additional amount of 0.5 mM NaBH4, with stirring. Following the reduction, the samples were repeatedly centrifuged and washed with water to remove unreacted AgNO3 and HAuCl4. The samples were then dried at 80°C.
Photocatalytic degradation experiment
The photocatalysts, SiO2 NPs, SiO2 NPs doped with Ag+ or Au3+, and SiO2 NPs with Ag and/or Au NPs deposited on the surface, were added to a MR solution to obtain 100 ml of 500 mg l–1 photocatalyst and 50 ppm of MR. Then, to establish the adsorption equilibrium between the MR and the photocatalysts, the resulting solution was stirred in the dark for 20 min. The zero time reading was taken and the solution was exposed to a xenon lamp.
Instruments
The setup used for the photocatalytic degradation experiments consisted of a 250 ml beaker with a xenon lamp (50 W, Orial mode 66001), mounted 20 cm from the surface for use as a source of artificial sunlight. The solution was stirred during irradiation.
A Bio-carry 50 UV-Vis spectrophotometer, with a range of 190–1100 nm, was used to measure the absorption spectra of MR as a function of irradiation time. Transmission electron microscopy (TEM) (JEOL) was used to determine the particle size and morphology. Transmission electron microscopy images were taken by drying a drop of colloidal photocatalyst on a surface of a copper-coated carbon grid. Particle size and distribution were obtained from the 150 particles within the TEM image that were enlarged and measured.
The MR dye has an absorption maximum at 522 nm (pH ~4) and its concentration can be determined optically with the aid of a calibration curve. The results revealed a linear relationship for all concentrations of the dye (i.e. Beer's law was obeyed).
Results and discussion
Determination of the particle size of silica photocatalysts
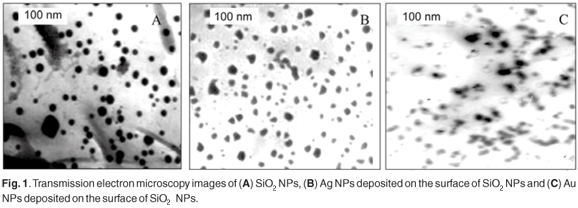
The catalytic power of such particles is strongly correlated to particle size and distribution of sizes. This is because the particle's size has a great effect on the energy levels of the nano-photocatalyst. Consequently, to obtain a good understanding of the photocatalytic processes, the particle size, morphology, and size distribution should be studied. Figure 1 represents the TEM image of (a) SiO2 NPs and (b) Ag NPs or (c) Au NPs deposited on the surface of SiO2 NPs. The SiO2 NPs were spherical with a large size distribution (15 ± 8 nm). The Ag NPs and Au NPs deposited on the surface of SiO2 NPs had an irregular shape due to the presence of particles. The size distributions were 17 ± 5 nm and 19 ± 6 nm for Ag NPs and Au NPs, respectively.
Photocatalytic degradation of MR dye by SiO2 NPs
The absorption band of MR dye was found to be centred at 522 nm. No change in the optical absorption peak intensity was observed in the presence or absence of SiO2 NPs, for up to 10 h in the dark. Additionally, only a very small change in the intensity of the maximum absorption was observed after 120 min irradiation in the absence of SiO2 NPs.
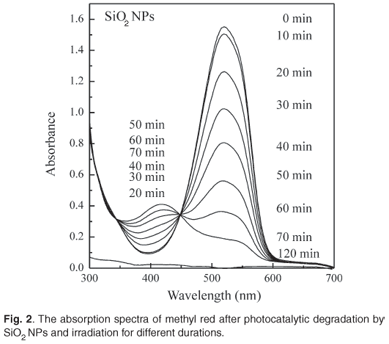
Figure 2 shows the absorption spectra of MR before and after exposure to the xenon lamp for different lengths of time. In the presence of SiO2 NPs as a photocatalyst, the intensity of the peak was found to decrease with increasing irradiation time due to photocatalytic degradation of MR. In addition to the first peak, a second peak appeared at about 415 nm and increased with time for 50 min, after which it decreased. Complete bleaching of the solution was obtained after 120 min.
Photocatalytic degradation of MR dye by Ag NPs and Au NPs deposited on the surface of SiO2 NPs
The Ag NPs and Au NPs had a great effect on the catalytic properties of SiO2 NPs. The photocatalytic degradation of MR was observed in the presence of Ag NPs and/or Au NPs deposited on the surface of SiO2 NPs. The band corresponding to MR absorption was found to decrease with time in the case of both pure Ag NP- and pure Au NP-deposited SiO2 NP samples. The rate of decay was sharper in the case of the Ag NPs. Both Ag NPs and Au NPs had rates of decay greater than pure SiO2 NPs. The peak rate of decay of MR, in the case of co-deposited Au NPs and Ag NPs on SiO2 NPs, lay between the rates of decay for pure Ag NPs and pure Au NPs. Complete discoloration took place after 70 min, 90 min and 75 min for Ag NPs, Au NPs, and Au NPs and Ag NPs co-deposited on SiO2 NPs, respectively. It was found that the concentration of intermediate products was lower in the case of Au NPs compared to Ag NPs, but these products decreased after 35 min in both cases.
Photocatalytic degradation of MR dye by SiO2 NPs doped with Ag+ and Au3+ ions
The photocatalytic degradation of MR by SiO2 NPs was found to be enhanced in the presence of Ag+ or Au3+ ions. The rate of MR degradation was found to be faster in Au3+ doped SiO2 NPs than when Ag+ was used. The colour of SiO2 NPs became dark in the case of Ag+ and Au3+ ions. The amount of intermediate products was found to increase with time in the case of doping with Ag+ and Au3+ until 20 min and 5 min into the reaction, respectively; it then decreased until complete discoloration at 60 min and 13 min, respectively.
Kinetics study of photocatalytic degradation
The Langmuir-Hinshelwood model30 can be used to describe the relationship between the rates of the photocatalytic degradation of dye in the presence of SiO2 NPs as a function of irradiation time. The rate equation is used in the form:31

where Kad is the adsorption coefficient of the reactant on SiO2, kL-H is the reaction rate constant and C is the concentration at any time t. The values of kL-H and Kad are used to explain the effect of light intensity on the equilibrium constant for the fast adsorption-desorption processes between the surface monolayer at SiO2 and the bulk solution. Then, by integration of Equation 1:
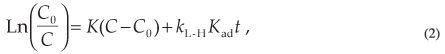
where C0 is the initial concentration.
For pseudo-first-order reaction KadC is very small compared to 1 in the denominator of Equation 1, so it is simplified and integrated to be:

where k = kL–H Kad is the pseudo-first-order reaction rate constant, and the half-life time t(1/2) can be calculated using the following expression:

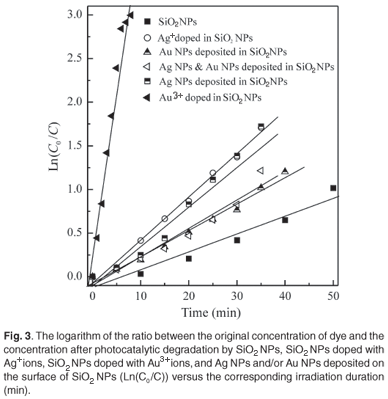
Plotting the natural logarithm of the ratio between the original concentration of MR and the concentration after photocatalytic degradation (Ln(C0/C) versus the corresponding irradiation time (min) yields a linear relationship as shown in Fig. 3. Therefore, the photocatalytic degradation reaction of MR by SiO2 NPs belongs to the pseudo-first-order reaction kinetics. The rate constant is the slope of the straight line in Fig. 3. Equation 4 was used to calculate the half-life time for the photocatalytic degradation of MR by SiO2 NPs, SiO2 NPs doped with Ag+ ions, SiO2 NPs doped with Au3+ ions, and Ag NPs (and/or) Au NPs deposited on the surface of SiO2 NPs. These values are summarised in Table 1.
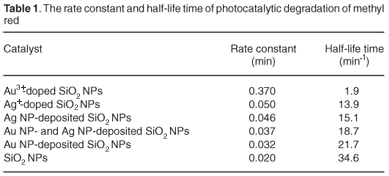
The mechanism for photocatalytic degradation of MR dye
Jiang et al.32 studied the negative shift in the X-ray photoelectron spectra of SiO2 NPs coated with Ag NPs compared to pure SiO2 NPs and reported that this shift could be attributed to the electron transfer between Ag NPs and SiO2 NPs. Moreover, SiO2 NPs were found to be photoexcited under UV irradiation and showed an absorption band at ~309 nm, which was attributed to the charge transfer from the bonding orbital of Si–O to the 2p nonbonding orbital of non-bridging oxygen.33
When a photon of UV light strikes the SiO2 surface, an electron from its valence band (vb) jumps to the conduction band (cb) leaving behind a positively charged hole (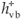 ). The negative charge is increased in the conduction band (
). The negative charge is increased in the conduction band ( 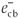 ) and photocatalytic active centres are formed on the surface of SiO2 NPs (as proven from the photoluminescence measurements)20 according to Equation 5:
) and photocatalytic active centres are formed on the surface of SiO2 NPs (as proven from the photoluminescence measurements)20 according to Equation 5:

The valence band holes react with the chemisorbed H2O molecules to form reactive species such as ·OH radicals, which subsequently react with dye molecules to cause their complete degradation.
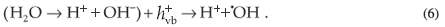
Alternatively, and can recombine on the surface of the particle within a few nanoseconds and the resulting energy is dissipated as heat. Furthermore, the and the can be trapped in surface states where they may react with species adsorbed or close to the surface of the particle. The can react with an acceptor, such as dissolved O2, which consequently is transformed into a super oxide radical anion (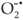 ) which leads to the formation of additional
) which leads to the formation of additional 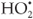 in Equation 7.
in Equation 7.
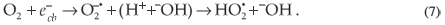
On the other hand, , could interact with donors, such as –OH and ·O2H, to form ·OH radicals. These radicals also attack the MR in the following manner:

The main factor affecting the efficiency of SiO2 NPs is the amount of ·OH radicals as described above. Therefore, any factor that supports the generation of ·OH radicals will enhance the rate of photocatalytic degradation of MR. When Au3+ or Ag+ ions are present during the reaction, they are absorbed on the surface of SiO2 NPs and then combine with the electrons in the conduction band of SiO2 NPs to form the corresponding metal (Equation 9). These ions reduce the recombination of charges ( and ) and favour the formation of ·OH radicals.34 The enhancing effect of Au3+ and Ag+ may be explained by their ability to trap electrons and generate holes so they act as electron scavengers.35
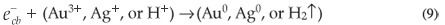
The mechanism of photocatalytic degradation of MR by the surface of SiO2 NPs loaded with Ag NPs, Au NPs, or both Au and Ag NPs could be controlled by the deposited nanoparticles because of their effect on the electron-hole recombination process.36 The major role of the deposited surface particles is the consumption of electrons and passing of those electrons to H+ ions or to O2. The retardation of the electron-hole recombination will increase the photocatalytic efficiency of the SiO2 NPs photocatalysts and, consequently, accelerate hydroxyl radical formation which will enhance the rate of MR degradation,37,38 Equations 10–12:
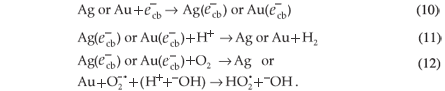
The mentioned mechanism accorded well with the experimental results. The most efficient catalyst was SiO2 NPs doped with Au3+ ions, because each Au3+ ion can consume three and generate three ·OH ions which are responsible for degradation of the dye. The SiO2 NPs doped with Ag+ ions were less efficient due to the generation of only one ·OH by each Ag+ ion.
Au NPs or Ag NPs deposited on the surface of SiO2 NPs act as electron–hole separation centres.39,40 Thermodynamically, the electron transfer from the SiO2 NP conduction band to the conduction band of the metal NPs, at the interface, occurs because the Fermi level of SiO2 is higher than that of Au NPs or Ag NPs.41 Consequently, a Schottky barrier at the Au NPs or Ag NPs SiO2 NPs contact region is formed, which improves the charge separation and enhances the photocatalytic activity of SiO2. The energy difference between the valence and conduction bands of Au NPs is lower than that of Ag NPs. This allows Ag NPs to increase the activity of SiO2 NPs more than do Au NPs.
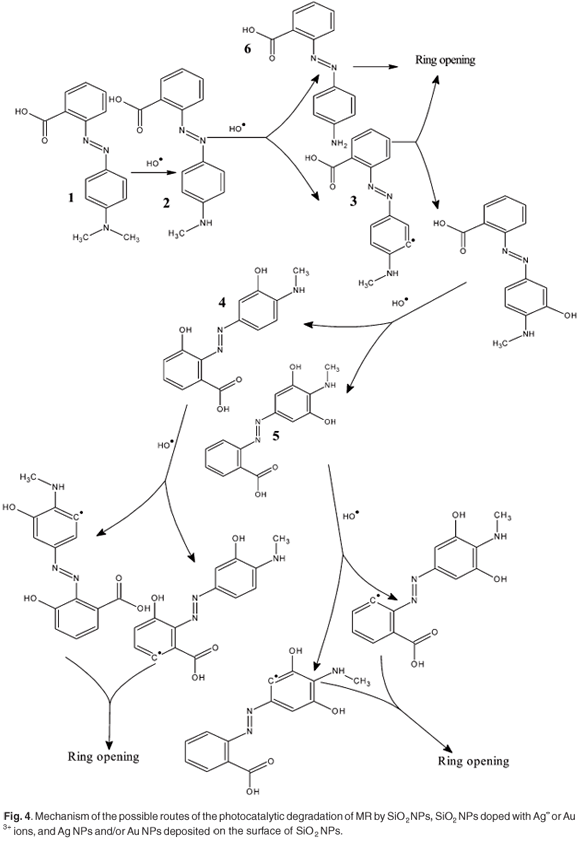
The hydroxide attack might be responsible for the discoloration mechanism of the MR dye, as summarised in the schematic diagram (Fig. 4). Including the proposed steps, the hydroxide radical is assumed to interact with MR or the intermediate photoproducts as follows: first the ·OH attacks MR which leads to formation of a dehydrogenated radical (intermediate 2). From this point, intermediate 2 can either undergo ring opening or combination with ·OH, forming hydroxyl product 3. The hydroxyl product causes the broadening in the absorption spectra of the remaining MR, as shown in Fig. 2. The intermediate 2 might also be decomposed to form a new low molecular weight byproduct, which has a blue-shifted absorption compared to the MR monomer (λmax. ~ 415 nm).42,43 This peak, which might correspond to a low molecular weight byproduct formation, was found to increase as the rate of the photocatalytic reaction decreased (decreasing the catalytic power of silica as shown in the profile of pure silica). Product 3 can be further attacked by ·OH to form bi-hydroxyl products 4 and 5 or a dehydrogenated radical which undergoes ring opening. The same procedure could take place for products 4 and 5 until complete ring opening and final mineralisation.
1. Buitron C.L., Quezada M. and Moreno G. (2004). Aerobic degradation of the azo dye acid red 151 in a sequencing batch bio-filter. Bioresource Technol. 92, 143–149. [ Links ]
2. Sokmen M., Allen D.W., Akkas F., Kartal N. and Acar F. (2001). Photodegradation of some dyes using Ag-loaded titanium dioxide. Water Air Soil Pollut. 132, 153–163. [ Links ]
3. Sauer T., Nero G.C., Jose H.J. and Moreira R.F.P.M. (2002). Kinetics of photocatalytic degradation of reactive dyes in a TiO2 slurry reactor. J. Photochem. Photobiol. A: Chem. 149, 147. [ Links ]
4. Fornasari G. and Trifiro F. (1998). Oxidation with no-redox oxides: ammoximation of cyclohexanone on amorphous silicas. Catal. Today 41, 443–455. [ Links ]
5. Bal R., Tope B.B., Das T.K., Hegde S.G. and Sivasanker S. (2001). Alkali-loaded silica, a solid base: investigation by FTIR spectroscopy of adsorbed CO2 and its catalytic activity. J. Catal. 204, 358–363. [ Links ]
6. Haber J., Pamin K., Matachowski L. and Mucha D. (2003). Catalytic performance of the dodecatungstophosphoric acid on different supports. Appl. Catal. A 256, 141–152. [ Links ]
7. Yoshida H., Chaskar M.G., Kato Y. and Hattori T. (2003). Active sites on silica-supported zirconium oxide for photoinduced direct methane conversion and photoluminescence. J. Photochem. Photobiol. A: Chem. 160, 47–53. [ Links ]
8. Yuliati L., Hattori T. and Yoshida H. (2005). Highly dispersed magnesium oxide species on silica as photoactive sites for photoinduced direct methane coupling and photoluminescence. Phys. Chem. Chem. Phys. 7, 195–201. [ Links ]
9. Yoshida H., Matsushita N., Kato Y. and Hattori T. (2003). Synergistic active sites on SiO2–Al2O3–TiO2 photocatalysts for direct methane coupling. J. Phys. Chem. B 107, 8355–8362. [ Links ]
10. Badr Y., Abd El-Wahed M.G. and Mahmoud M.A. (2008). Photocatalytic degradation of methyl red dye by silica nanoparticles. J. Hazard. Mater. 154, 245–253. [ Links ]
11. Yoshida H., Shimizu T., Murata C. and Hattori T. (2003). Highly dispersed zinc oxide species on silica as active sites for photoepoxidation of propene by molecular oxygen. J. Catal. 220, 226–232. [ Links ]
12. Ogata A., Kazusaka A. and Enyo M. (1986). Photoactivation of silica gel with UV light during the reaction of carbon monoxide with oxygen. J. Phys. Chem. 90, 5201–5205. [ Links ]
13. Inaki Y., Yoshida H. and Hattori T. (2000). Two photoexcitation steps for photometathesis of propene over FSM-16. J. Phys. Chem. B 104, 10304–10309. [ Links ]
14. Yoshida H., Kimura K., Inaki Y. and Hattori T. (1997). Catalytic activity of FSM-16 for photometathesis of propene. Chem. Commun. 129–130. [ Links ]
15. Yoshida H., Matsushita N., Kato Y. and Hattori T. (2002). Active sites in sol-gel prepared silica-alumina for photoinduced non-oxidative methane coupling. Phys. Chem. Chem. Phys. 4, 2459–2467. [ Links ]
16. Yoshida H., Kato Y. and Hattori T. (2000). Photoinduced non-oxidative methane coupling over silica-alumina. Stud. Surf. Sci. Catal. 130, 659–664. [ Links ]
17. Yoshida H., Chaskar M.G., Kato Y. and Hattori T. (2002). Fine structural photoluminescence spectra of silica-supported zirconium oxide and its photoactivity in direct methane conversion. Chem. Commun. 2014–2015. [ Links ]
18. Kato Y., Matsushita N., Yoshida H. and Hattori T. (2002). Highly active silica-alumina-titania catalyst for photoinduced non-oxidative methane coupling. Catal. Commun. 3, 99–103. [ Links ]
19. Yoshida H., Murata C. and Hattori T. (2000). Screening study of silica-supported catalysts for photoepoxidation of propene by molecular oxygen. J. Catal. 194, 364–372. [ Links ]
20. Inaki Y., Yoshida H., Yoshida T. and Hattori T. (2002). Active sites on mesoporous and amorphous silica materials and their photocatalytic activity: an investigation by FTIR, ESR, VUV–UV and photoluminescence spectroscopies. J. Phys. Chem. B 106, 9098–9106. [ Links ]
21. Furukawa T., Fox K.E. and White W.B. (1981). Raman spectroscopic investigation of the structure of silicate glasses. III. Raman intensities and structural units in sodium silicate glasses. J. Chem. Phys. 75, 3226–3237. [ Links ]
22. Skuja L. (2000). Optical Properties of Defects in Silica, eds G. Pacchioni, L. Skuja and D.L. Griscom, pp. 73–116. Kluwer Academic Publishers, the Netherlands. [ Links ]
23. San N., Hatipoglu A., Kocturk G. and Cmar Z. (2002). Photocatalytic degradation of 4-nitrophenol in aqueous TiO2 suspensions: theoretical prediction of the intermediates. J. Photochem. Photobiol. A: Chem. 146, 189–197. [ Links ]
24. Siemon U., Bahnemann D., Testa J.J., Rodriguez D., Litter M.I. and Bruno N. (2002). Heterogeneous photocatalytic reactions comparing TiO2 and Pt/TiO2. J. Photochem. Photobiol. A: Chem. 148, 247–255. [ Links ]
25. Ohtani B., Iwai K., Nishimoto S. and Sato S. (1997). Role of platinum deposits on titanium(IV) oxide particles: structural and kinetic analyses of photocatalytic reaction in aqueous alcohol and amino acid solutions. J. Phys. Chem. B. 101, 3349–3359. [ Links ]
26. Gerischer H. and Heller A. (1991). The role of oxygen in photooxidation of organic molecules on semiconductor particles. J. Phys. Chem. 95, 5261–5267. [ Links ]
27. Badr Y. and Mahmoud M.A. (2007). Photocatalytic degradation of methyl orange by gold silver nano-core/silica nano-shell. J. Phys. Chem. Solids 68, 413–419. [ Links ]
28. Subba Rao K.V., Lavedrine B. and Boule P. (2003). Influence of metallic species on TiO2 for the photocatalytic degradation of dyes and dye intermediates. J. Photochem. Photobiol. A: Chem. 154, 189–193. [ Links ]
29. Kamat P.V., Flumiani M. and Dawson A. (2002). Metal–metal and metal–semiconductor composite nanoclusters. Colloids and Surf. A 202, 269–279. [ Links ]
30. Houas A., Lachheb H., Ksibi M., Elaloui E., Guillard C. and Herrmann J. (2001). Photocatalytic degradation pathway of methylene blue in water. Appl. Catal. B: Environ. 31, 145–157. [ Links ]
31. Sun Z., Chen Y., Ke Q., Yang Y. and Yuan J. (2002). Photocatalytic degradation of a cationic azo dye by TiO2/bentonite nanocomposite. J. Photochem. Photobiol. A: Chem. 149, 169–174. [ Links ]
32. Jiang Z., Liu C. and Liu Y. (2004). Formation of silver nanoparticles in an acid-catalyzed silica colloidal solution. Appl. Surf. Sci. 233, 135–140. [ Links ]
33. Skuja L. (1992). Time-resolved low temperature luminescence of non-bridging oxygen hole centers in silica glass. Solid State Commun. 84, 613–616. [ Links ]
34. Fu G., Cai W., Gan Y. and Jia J. (2004). An ambience-induced optical absorption peak for Au/SiO2 mesoporous assembly. Chem. Phys. Lett. 385, 15–19. [ Links ]
35. Yang Z.X., Wu R.Q. and Goodman D.W. (2000). Critical behavior of ac conductivity near the Anderson transition. Phys. Rev. B 6, 14066–14071. [ Links ]
36. Hirakawa T. and Kamat P.V. (2005). Charge separation and catalytic activity of AgTiO2 core-shell composite clusters under UV-irradiation. J. Am. Chem. Soc. 127, 3928–3934. [ Links ]
37. Sakthivel S., Shankar M.V. and Palanichamy M. (2004). Enhancement of photocatalytic activity by metal deposition: characterisation and photonic efficiency of Pt, Au and Pd deposited on TiO2 catalyst. Water Res. 38, 3001–3008. [ Links ]
38. Linsbigler A., Rusu C. and Yates J.T. (1994). Absence of platinum enhancement of a photoreaction on TiO2–CO photooxidation on Pt/TiO2(110). J. Am. Chem. Soc. 118, 5284–5289. [ Links ]
39. Henglein A. (1979). Reactions of organic free radicals at colloidal silver in aqueous solution. Electron pool effect and water decomposition. J. Phys. Chem. 83, 2209–2216. [ Links ]
40. Herrmann J.M., Disdier J. and Pichat P. (1986). Photoassisted platinum deposition on TiO2 powder using various platinum complexes. J. Phys. Chem. 90, 6028–6034. [ Links ]
41. Sclafani A. and Herrmann J.M. (1998). Influence of metallic silver and of platinum-silver bimetallic deposits on the photocatalytic activity of titania (anatase and rutile) in organic and aqueous media. J. Photochem. Photobiol. A: Chem. 113, 181–188. [ Links ]
42. Comparelli R., Fanizza E., Curri M.L., Cozzoli P.D., Mascolo G. and Agostiano A. (2005). UV-induced photocatalytic degradation of azo-dyes by organic-capped ZnO nanocrystals immobilized onto substrates. Appl. Catal. B: Environ. 60, 1–11. [ Links ]
43. Comparelli R., Cozzoli P.D., Curri M.L., Agostiano A. and Mascolo G. (2004). Surface diagnostics for scale analysis. Water Sci. Tech. 49, 183–190. [ Links ]
Received 18 May. Accepted 27 July 2009.
* Author for correspondence E-mail: mahmoudchem@yahoo.com
